Theoretical Study of the Triplet Electronic States of the BP Molecule ()
1. Introduction
Although the molecular III-V family have many unique chemical and physical properties, boron phosphate has a good translucent, good adhesion, smaller index of ionicity and low internal stress level. It also, constitutes an excellent example of an almost perfect covalence heteronuclear diatomic molecule. This low-heteropolarity gives striking features to the BP system. Many experimental studies were done on diatomic molecules by considering either one electronegative and one electropositive atom or two electronegative atoms. From the other side, no extensive studies were done on diatomic molecules of two atoms belonging to electropositive groups [1] [2] . The boron phosphate has a wide range of applications in refractory materials and solid state physics, and in a new kind of infrared optical materials [3] [4] .
In literature, BP molecule was originally studied by Gingerich et al. [5] . Later, Boldyrev and Simons [6] investigated 5 electronic states for the molecule BP. They conclude that the 3P state is the ground state, while the first excited state 1S+ lies 0.3 eV above the ground state. Since the BP molecule has very close electronic states of different multiplicity, there are difficulties for determining its molecular parameters in the fundamental and excited states. Using Bader’s theory of atoms in molecules [7] [8] , BP compound, in both crystalline and molecular structure, was studied by Mori-Sánchez et al. [9] -[11] . He analyzed the transfer of charge and the polarity inversion under pressure in the electron density of the BP crystal. Mori-Sánchez [11] has analyzed the electron density of the diatomic BP molecule at different computational levels, namely HF, CASSCF and CISD and he found that, a competition between the three states of lower energy 1S+, 3S−, and 3P and concluded that 3P is the fundamental state in agreement with the results of Boldyrev and Simons [6] . More theoretical calculations have been done using different techniques as HF, CASSCF, CCSD and FCI [12] -[16] where few spectroscopic constants for the low excited electronic states and the dissociation equilibrium have been obtained.
The focus of this study is devoted to an accurate description of the ground and electronically excited triplet electronic states. In order to emphasize the accuracy of our work, we used the multi-reference configuration interaction (MRCI+Q) model expansion. The spectroscopic constants Re, Te, Be, ωe, …for each of the corresponding electronic states have been investigated along with the static dipole moment.
2. Method of Calculations
The correlation energy for a certain state is the difference between the exact eigenvalue of the Hamiltonian and its expectation value in the HF approximation. The configuration interaction (CI) treatment of this electron correlation is obtained by adding to the HF wavefunction terms that represent promotion of electrons from occupied to virtual (unoccupied) orbital that are singly, doubly, and triply excited. Each Slater determinant D, or linear combination of a few determinants, represent an idealized configuration, called configuration state function (CSF). The CI wavefunctions then, are linear combinations of CSFs. The most widely-used implementations of CI are multiconfigurational self consistent field (MCSCF) method. In this method, one writes the molecular wavefunction as a linear combination of CSFs and varies not only the expansion coefficients but also the forms of the molecular orbitals in the CSFs. CASSCF wavefunctions are often used as the starting point for MRCI calculation. When a CASSCF wavefunction is used for a MRCISD calculation, the number of CSFs may be too many to deal with, so one procedure used to reduce the amount of computations is the internally contracted MRCI. The theoretical study of the low-lying singlet and triplet electronic states of the molecule BP have been studied by using the state averaged Complete Active Space Self-Consistent Field (CASSCF) procedure [17] [18] followed by a Multireference Doubly and Singly Configuration Interaction MRDSCI+Q with Davidson correction [19] -[22] . The entire CASSCF configuration space was used as the reference in the MRDSCI calculations, which were done via the computational chemistry Program MOLPRO [23] taking advantage of the graphical user interface GABEDIT [24] . This software is intended for high accuracy correlated ab initio calculations. MOLPRO has been run on a PC-computer with LINUX-type operating systems. The boron and phosphorous species are treated in all electron scheme. The basis sets were chosen for s, p and d functions using aug-cc-pCVTZ; c and aug-cc- pVTZ; c for B and P atoms respectively from Molpro library. The CASSCF active space is 6s (B: 2p, 3s; P:3p0, 4s, 3d0, 4p0) and 4p (B:2p±1; P:3p±1, 3d±1, 4p±1) 1d (P:3d±2) orbitals in the C2v molecular orbitals are distributed into the irreducible representation a1, b1, b2 and 1a2 in the following way 6a1, 4b1, 4b2, 1a2 noted [6, 4, 4, 1]. The 1s2 2s2 of B and 1s2 2s2 2p6 3s2 of P atoms were frozen in the MCSCF procedure. The number of active orbitals and valence electrons are 16 and 8 respectively.
3. Results and Discussion
Potential energy curves (PECs) of the 17 triplet electronic states, in the representation 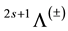 of the molecule BP, has been performed for 189 internuclear distances. These curves are given in Figure 1.
of the molecule BP, has been performed for 189 internuclear distances. These curves are given in Figure 1.
One can notice the deep potential wells for the low doublet and shallower wells for the higher excited electronic states. Moreover, some crossings and avoided crossings of abscissas Rc and Rac respectively have been obtained between the potential energy curves. If the two curves correspond to states of different symmetry cross where the crossing in this case is strictly allowed. But if these wavefunctions have the same symmetry, the two
![]()
Figure 1. Potential energy curves of the triplet electronic states of the molecule BP.
states will only be diabatic solutions of the problem. They will mix with each other to give two adiabatic solutions which no longer cross and the crossing becomes avoided. The adiabatic solutions of the Schrödinger equation Y1 and Y2 are obtained by linear combinations of the diabatic ones where the variation method is used to solve for the coefficients. Such crossings or avoided crossings can dramatically alter the stability of molecules and the trend of the potential energy curve. From Figure 1, we can find these avoided crossings between the electronic states (1)3D/(2)3D at 2.23 Å and (2)3D/(3)3D at 2.77 Å. More over the wells are absent from some states which are unbound states. The electronic states (2)3P presents double wells where we calculated the spectroscopic constants in Table 1. The potential energy curves for the 17 low-lying triplet electronic states of the molecule BP have been calculated using the MRSDCI method in the representation 2s+1Λ(±). These curves are drawn versus the internuclear distance 1.2 Å ≤ R ≤ 5.5 Å in Figure 1. The equilibrium bond distances Re, the harmonic vibrational frequencies ωe, the relative energy separations Te, and the rotational constants Be for these electronic states have been calculated and given in Table 1.
The comparison of our calculated internuclear distance Re for the ground state X3P with those given in literature, either experimental or theoretical, shows a very good agreement with relative difference of 0.4% (Ref. [16] ) ≤ DRe/Re ≤ 1.4% (Ref. [18] ). Similar very good agreement is obtained by comparing our calculated value of Re,
![]()
Table 1. Spectroscopic constants of the potential wells of the electronic states of the BP molecule.
aPresent theoretical study. bRef [15] , cRef [16] , dRef [17] , eRef [18] , fRef [6] , gRef [12] .
for the first excited state (1)3Σ−, with those given in literature where the relative difference is 1.5% (Ref. [16] ) ≤ DRe/Re ≤ 2.1% (Ref. [15] ), while this relative difference becomes DRe/Re = 7.7% for the state (2)3Π.
One can notice the large discrepancy between the given values for the harmonic frequency ωe in literature, but our calculated value is found to be between the highest and lowest values as 934.69 cm−1 < 1026.1 cm−1 < 1148 cm−1. While our calculated value of we is greater than those given in literature for the electronic state (1)3Σ− and smaller for the state (2)3Π. The investigated values of the rotational constant Be in literature are in very good agreement with our calculated values for the 2 electronic states X3Π and (1)3Σ− and larger for the (2)3Π state. The calculated value of the energy Te with respect to the ground state is in an acceptable agreement with the theoretical data given in literature. From this agreement between our investigated data and those calculated in literature we can pretend the accuracy of the new constants obtained for these investigated new electronic excited states.
4. Permanent Dipole Moment
The electric dipole moment function Re(r) of a molecule is given by:
 (1)
(1)
where  and
and 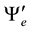 are respectively the electronic wavefunctions of the two different electronic states and
are respectively the electronic wavefunctions of the two different electronic states and 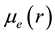 is the permanent dipole moment. This permanent dipole moments is investigated for the considered electronic states of the molecule BP by taking the boron (B) atom at the origin, and the phosphorus (P) atom moves along the positive z-axis. All the calculations were performed with the MOLPRO [23] program. The dipole moment operator is among the most reliably predicted physical properties, because the quantum mechanical operator is a simple sum of one-electron operators. The expectation value of this operator is sensitive to the nature of the least energetic and most chemically relevant valence electrons. The positive sign of the dipole moment corresponds to a charge transfer from the P atom towards the B atom. To obtain the best accuracy, MRCI wavefunctions were constructed using MCSCF active space. The values of the dipole moments for the investigated electronic states are given in atomic unites (a.u.) as a function of the internuclear distance R in Figure 2.
is the permanent dipole moment. This permanent dipole moments is investigated for the considered electronic states of the molecule BP by taking the boron (B) atom at the origin, and the phosphorus (P) atom moves along the positive z-axis. All the calculations were performed with the MOLPRO [23] program. The dipole moment operator is among the most reliably predicted physical properties, because the quantum mechanical operator is a simple sum of one-electron operators. The expectation value of this operator is sensitive to the nature of the least energetic and most chemically relevant valence electrons. The positive sign of the dipole moment corresponds to a charge transfer from the P atom towards the B atom. To obtain the best accuracy, MRCI wavefunctions were constructed using MCSCF active space. The values of the dipole moments for the investigated electronic states are given in atomic unites (a.u.) as a function of the internuclear distance R in Figure 2.
At large internuclear distances, the dipole moment of all the investigated electronic states either tends to a small constant indicating a nearly covalent character of the corresponding state or it smoothly approaches zero which is theoretically the correct behavior for a molecule that dissociates into natural fragments.
5. Conclusion
In the present work, ab initio investigation for 17 low-lying electronic states in the representation 2s+1Λ(+/−) of the
![]()
Figure 2. Dipole moment curves of the triplet electronic states of the molecule BP.
BP molecule was computed via CASSCF/MRSDCI methods for the electronic excited states. The potential energy curves, the electronic energy with respect to the ground state Te, the harmonic frequency ωe, the internuclear distance Re have been calculated along with the rotational constant Be. The comparisons of the present results with the available values in the literature show an overall a very good agreement. To the best of our knowledge, 14 new electronic states have been investigated for the first time through this work. With the recent interest on this molecule through, the present study of these new excited electronic states may assist in searching for bound states of the BP molecule, which leads to more investigation of new experimental works on this molecule.
NOTES
*Corresponding author.