Recent Advances in X-Ray Structures of Metal-Phenoxyl Radical Complexes ()
1. Introduction
The oxidation chemistry of metal complexes has been widely developed in recent years, affording deep insights into the reaction mechanisms for many useful homogeneous catalytic reactions and reactions at the active site of metalloenzymes [1].
Some oxidative reaction intermediates have been successfully characterized by various methods [2-11], and especially X-ray crystal structure analysis of some intermediates revealed the geometrical change in the reaction and the oxidant binding structure of metal complexes [5-12]. On the other hand, the oxidation state of the metal ions in the active species has not been fully understood, because the oxidation locus on oxidized metal complexes is often different from the “formal” oxidation site [13-15]. The “formal” and “experimental” oxidation numbers are frequently used as synonyms, since the term of the physical or experimental oxidation state has not been accepted in some areas of chemistry.
The valence state difference began to attract attention in early 1990’s from the studies on galactose oxidase (GO) [16-18]. GO is a single copper oxidase, which catalyzes a two-electron oxidation of a primary alcohol to the corresponding aldehyde. The active site structure of the inactive form of GO is shown in Figure 1, where two imidazole rings of histidine residues, two phenol moie-

Figure 1. Active site structure of the inactive form of GO.
ties of tyrosine residues, and an acetate ion are coordinated to the copper(II) ion [19].
Conversion to the active form of GO occurs upon one-electron oxidation, since this active form should act as a two-electron oxidant [16-19]. The formal oxidation state of the active form of GO can be described as a Cu(III)-phenolate species. Actually, the active form of GO had been considered to be a copper(III) species [20, 21], but various spectroscopic studies of the active form of GO revealed the formation of the phenoxyl radical species and the Cu(II)-phenoxyl radical bond [22]. The free phenoxyl radical is very unstable with the half life estimated to be 2.4 μs under ambient conditions, while the Cu(II)-phenoxyl radical in the active form of GO has a long life; the radical is not quenched for more than one week at room temperature [23]. Thus, properties of the metal coordinated phenoxyl radical show a significant change from those of the free phenoxyl radical.
The valence state difference as expected for one-electron oxidized Cu(II)-phenolate species can be detected in the X-ray crystal structure. For example, the formal oxidation number of the central metal ion of the complexes of iminophenolate dianion, (LAP)2− is not always identical with the experimental valence state [24,25]. In the case of [Ni(LAP)2]0, the formal oxidation state of the central nickel ion is +IV, but the experimental valence state of nickel can be assigned to +II, and two iminophenolate dianions are oxidized to iminosemiquinonate radical anions (LSQ)− (Figure 2) [24]. The X-ray structure of this complex revealed that the electronic structure of the ligand is different from that of the free ligand, H2LAP; the C-O bond in complexes is shorter and the ortho-C-C bonds are longer in comparison to H2LAP, which is assignable to the semiquinonate monoradical. Findings on such an oxidation state difference were reported early in 2000, though some analogues of this complex had previously been reported [15]. One of the reasons for this may be considered to be that the X-ray structures of a series of these analogues did not show significant geometrical changes of the complexes as a whole. However, subtle ligand structural changes were already pointed out in the previous analyses [25].
This review deals with recent advances in the chemistry related with the oxidation behavior of some metalphenolate complexes and their properties, with emphasis on X-ray crystal structures of metal-phenoxyl radical complexes together with high valent metal-phenolate complexes. In general, the X-ray diffraction method is sometimes difficult for full understanding of the experimental
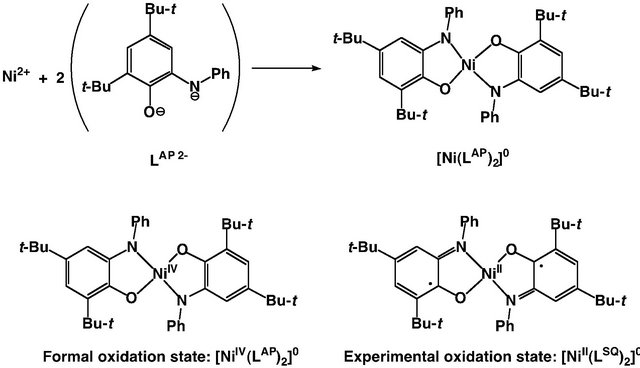
Figure 2. An example showing the difference between the experimental oxidation state and the formal oxidation number in the Ni complexes of iminophenolate dianions, [Ni (LAP)2]0.
valence state and the detailed electronic structure, while the recent high resolution analysis possibly reveals the electronic structures, since the small geometrical change upon oxidation can be detected. This review mainly focuses on the relationship between geometrical and electronic structures of one-electron oxidized metal-phenolate complexes.
2. Formation of Metal-Phenoxyl Radical Complexes
Due to the low stability of the uncoordinated free phenoxyl radical, the metal-phenoxyl radical complexes were prepared by one-electron oxidation of the metal-phenolato complexes. The first successful formation of the phenoxyl radical complexes was achieved by chemical and photochemical oxidations of the iron(III)-phenolato complexes (Scheme 1) [26].
This iron-phenoxyl radical complex was stable for more than one year under the dry air at room temperature, and therefore this complex could be isolated as a powder. The oxidants used for generation of the metal-phenoxyl radical species were unstable monoradical species, such as SO4•–. Use of more stable chemical reagents and electrochemical oxidation later became popular partly due to easy stochiometric handling. For these methods, determination of the redox potential of the radical formation is important. The tyrosyl radical formation potential by one-electron oxidation of the phenol moiety was reported to be 0.94 V vs. NHE [27], which is relatively high as compared with the stable metal-centered oxidation potentials such as those for FeII/FeIII and CoII/ CoIII [28]. The oxidation potentials of the metal coordinated phenolates so far reported are generally lower than those of free phenols, and the potential for generation of the active form of GO is as low as 0.41 V [21]. This may be due to the coordination effect of the metal ion. In 1996, Tolman and coworkers reported the CuII-phenoxyl radical complexes with similar ligands, which were obtained from the CuII-phenolate complexes by electrochemical oxidation and chemical oxidation using oneelectron oxidants such as (NH4)2Ce(NO3)6 (cerium(IV) ammonium nitrate, CAN) (Scheme 2) [29].

Scheme 1. The iron(III)-phenoxyl radical complex generated by photochemical and chemical oxidations from the iron(III)-phenolate complex.
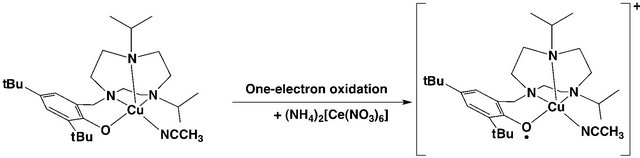
Scheme 2. The copper(II)-phenoxyl radical complex generated by addition of chemical oxidant (NH4)2Ce(NO3)6 (CAN) from the copper(II)-phenolate complex.
CAN is one of the strong oxidants with the CeIV/CeIII potential higher than 1.7 V, which indicates that it can fully oxidize metal-phenolato complexes by one electron to form the corresponding higher valent species [28]. Due to its stability and high potential this oxidant can be used stoichiometrically. On the other hand, CAN is soluble only in relatively polar solvents such as acetone and CH3CN, and it cannot be used for oxidation of metalphenolato complexes dissolved in CH2Cl2. In such cases stoichiometric addition of CAN is attained by adding a small volume of an over 10 times more concentrated solution of CAN in CH3CN to the CH2Cl2 solution of metal-phenoxyl radical complexes. Stack and coworkers reported formation of the radical species by stoichiometric addition of AgSbF6 [30]. This oxidant is soluble in CH2Cl2, forming silver(0) in the course of oxidation. The oxidation potential of AgI/Ag0 is 0.799 V vs. NHE, which is higher than the potentials for many metal-coordinated radicals [28,31]. After the reaction, this oxidant can be removed from the reaction mixture by filtration and is therefore useful for in situ crystallization of the metalphenoxyl radical species.
Elelctrochemical oxidation of the metal-phenolate precursors gives the metal-phenoxyl radical complexes [29], but the bulk electrolysis sometimes takes a long time in comparison to the life-time of the oxidized complex. On the other hand, generation of the metal-phenoxyl radical by air-oxidation has been reported [15,32], especially for the complexes of bidentate ligands, such as o-aminophenolate and catecholate, to give semiquinonate complexes [15]. However, the air-oxidation method is quite limited due to its low potential for the monodentate phenolate oxidation. Methods based on spontaneous formation of the metal-phenoxyl radical have been reported, formation by disproportionation of the Cu(II)-phenolate species [33, 34] being a unique example.
3. General Structural Features of Metal-Phenoxyl Radical Complexes
Free phenoxyl radicals are electron deficient and therefore highly reactive [35]. Even though specific techniques such as flash photolysis or pulsed radiolysis have been successfully used for detection of phenoxyl radicals, simple phenoxyl radicals are rather uncommon due to their low stability. Stabilization of the phenoxyl radicals has been attained by substitution of functional groups in the orthoand para-positions. The spin density of the radical electron on the phenoxyl radical species was estimated to be significantly high in orthoand para-positions. For understanding the electronic structure of the phenoxyl radical, the canonical presentation of phenoxyl radical is useful, as shown in Scheme 3. The canonical forms are reflected in the crystal structures of metal-phenoxyl radical complexes. The C-O bond length of the phenoxyl radical complexes becomes shorter and the ortho-C-C bonds are longer in comparison to the phenolate moiety. Further, π-electron donating substituents, such as the methoxy group, in orthoand para-positions also stabilize the phenoxyl radical, whose quinoid character is emphasized [36]. On the other hand, the metal-oxygen bond of the phenoxyl radical is longer than that of the metal-phenolate due to the electron deficiency of the phenoxyl radical. However, some of the phenoxyl radical complexes show the shortening of the metal-oxygen bond length of the phenoxyl radical as a result of delocalization of the unpaired electron (vide infra). In this case, no significant radical structural feature is shown in the crystal structure.
Difference between metal-centered oxidation and metalphenoxyl radical species can be detected in X-ray crystal structures. The metal-oxygen bond lengths of high-valent metal-phenolate complexes are shorter than those before oxidation, which is different from the characteristic of meta-phenoxyl radical complexes. The high-valent metalphenolate complexes should have “phenolate” moieties, so that the structural change of the phenolate moiety is rather small in comparison to the phenoxyl radical complexes [37].
4. Crystal Structures of Metal-Phenoxyl Radical Complexes
4.1. Cr(III)-Phenoxyl Radical Complex
Recently, X-ray crystal structures of some of the metalphenoxyl radical species have been reported, while the examples are still limited [37]. The crystal structures exhibit a geometry similar to those of the complexes before oxidation, but their details are different. Thus, the C-C and C-O bond lengths of the phenoxyl moiety were revealed to be different from the corresponding values of the phenolate complexes. The octahedral chromium(III) complex has a d3 configuration and half-filled t2g orbital in the central metal ion, so that the CrIII ion is paramag-

Scheme 3. Canonical forms of phenoxyl radical.
netic and redox innocent in the classical potential range. The CrIII-phenoxyl radical complex having a three phenolate moieties connected with the triazacyclononane backbone is the first example of the metal-monodentate phenoxyl radical complex revealed by the X-ray crystal structure analysis (Figure 3) [38]. The crystal structure of this complex having a p-methoxyphenoxyl radical shows a quinoid distribution of the bond lengths in the phenoxyl ring, as observed in the structure of the tri(alkyl)phenoxyl radical. As expected, the Cr-Ophenoxyl bond is weakened in comparison with the other phenolate moieties in this complex. Therefore, this complex is a relatively localized radical complex, which can be described as [Cr(phenoxyl)(phenolate)2]+. However, no X-ray crystal structures of the other metal complexes having this ligand and its analogues have been reported yet.
4.2. Crystal Structure of Cu-Phenoxyl Radical Complex
The first example of X-ray crystal structural analysis of the CuII-phenoxyl radical complex shown in Figure 4 has a relatively localized structure with one of the two phenolate moieties oxidized to the phenoxyl radical [39,40]. The two C-C bonds involving the C-O moiety in the radical are 0.03 - 0.04 Å longer and the C-O bond is ca. 0.06 Å shorter than the corresponding bonds in the phenolate moiety. The two Cu-O bond lengths are also different, the bond length for the phenoxyl radical being longer in comparison to the Cu-phenolate bond. Such characteristics are in good agreement with the corresponding structural features of the metal-phenoxyl radical complexes. Therefore, this complex has a localized electronic structure described as CuII(phenolate) (phenoxyl radical). Other metal complexes having this ligand, such as cobalt(II) and zinc(II) complexes, have also been reported. The Zn and Co complexes showed structural characteristics similar to those of this copper complex, indicating that they can also be assigned to the localized phenoxyl radical complexes, [MII(phenolate)(phenoxyl radical)]+. However, X-ray crystal structure analysis of the Co(II)-phenoxyl radical complex has not been reported yet [41].
5. Structures of One-Electron Oxidized Metal(II)-Shiff Base Complexes
5.1. Group 10 Metal Salen-Type Complexes (Metal = Ni(II), Pd(II) and Pt(II))
The X-ray crystal structure analyses of one-electron oxidized group 10 metal salen-type complexes are shown in Figure 5 [42-44]. The crystal structures of all these complexes were found to be similar to those of the corresponding complexes before oxidation, which supports the CV results that significant structure changes did not occur in the course of the oxidation.
However, a close look into the details of the structures

Figure 3. Molecular structure and selected bond lengths of the first metal-phenoxyl radical complex based on the X-ray crystal structure analysis; (A) molecular structure of the phenoxyl radical complex; (B), (C) Selected bond lengths of the phenolate (B) and phenoxyl radical (C) in the first phenoxyl radical complex based on the X-ray crystal structure analysis.

Figure 4. First example of the crystal structure of the CuII-phenoxyl radical complex. (A) crystal structure of the complex; (B), (C) Selected bond lengths of the phenolate ligand (B) and phenoxyl radical ligand (C) in the first Cu-phenoxyl radical complex based on the X-ray crystal structure analysis; (D) Canonical structures of the complex.
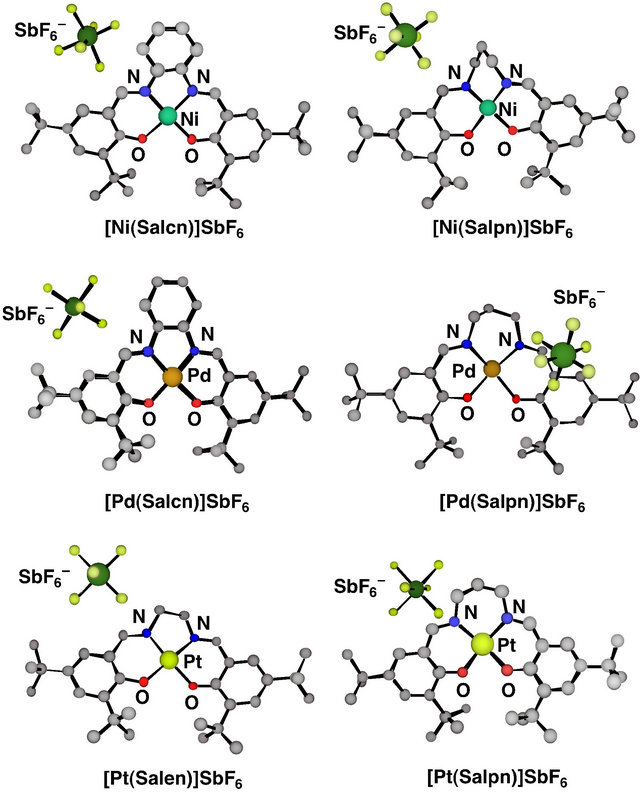
Figure 5. Ball and stick views of crystal structures of oneelectron oxidized group 10 metal salen-type complexes.
reveals that there are subtle differences between them, and especially the oxidized Pd(II) complexes are different from the other complexes [43]. Comparison of the 5-membered dinitrogen chelate backbones of the Salcn and Salen complexes indicates that upon oxidation the Ni and Pt complexes exhibited a clear coordination sphere contraction due to shortening of the M-O and M-N bond lengths (Table 1) [42,43]. On the other hand, the Pd complex showed an unsymmetrical contraction: One of the Pd-O bonds (2.003 Å) is longer than the other (1.963 Å), and the C-O bond (1.263 Å) of the phenolate moiety with the longer Pd-O bond is shorter than the other C-O bond (1.318 Å). The phenolate moiety with a shorter C-O bond length shows the lengthening of the ring ortho C-C bonds in comparison with the other C-C bonds (Table 1). These structural features of the phenolate moiety in the oxidized Pd complex are in good agreement with the characteristics of the phenoxyl radical, which is in the quinoid form due to delocalization of the radical electron on the phenoxyl ring as shown in Scheme 3. Such properties were also detected for the Pd complex with a 6-membered dinitrogen chelate back bone, [Pd(Salpn)] SbF6 [44]. In addition, the  counterion was positioned close to the quinoid moiety of this complex; the closest distance between the
counterion was positioned close to the quinoid moiety of this complex; the closest distance between the  and the C-O carbon atom of the phenoxyl ligand was 3.026 Å. Therefore
and the C-O carbon atom of the phenoxyl ligand was 3.026 Å. Therefore
Table 1. Comparison of the M-O and M-N, and C-C and C-O bond lengths (Å) in the phenolate moieties of the oxidized group 10 metal salen type complexes having a 5-membered dinitrogen chelate backbone.

aThe bond lengths are described for one of the two salen units of the molecule.
one-electron oxidized Pd(II) complexes can be assigned to relatively localized PdII(phenoxyl)(phenolate) complexes.
However, a close look into the details of the structures reveals that there are subtle differences between them, and especially the oxidized Pd(II) complexes are different from the other complexes [43]. Comparison of the 5-membered dinitrogen chelate backbones of the Salcn and Salen complexes indicates that upon oxidation the Ni and Pt complexes exhibited a clear coordination sphere contraction due to shortening of the M-O and M-N bond lengths (Table 1) [42,43]. On the other hand, the Pd complex showed an unsymmetrical contraction: One of the Pd-O bonds (2.003 Å) is longer than the other (1.963 Å), and the C-O bond (1.263 Å) of the phenolate moiety with the longer Pd-O bond is shorter than the other C-O bond (1.318 Å). The phenolate moiety with a shorter C-O bond length shows the lengthening of the ring ortho C-C bonds in comparison with the other C-C bonds (Table 2). These structural features of the phenolate moiety in the oxidized Pd complex are in good agreement with the characteristics of the phenoxyl radical, which is in the quinoid form due to delocalization of the radical electron on the phenoxyl ring as shown in Scheme 3. Such properties were also detected for the Pd complex with a 6-membered dinitrogen chelate back bone, [Pd(Salpn)] SbF6 [44]. In addition, the  counterion was positioned close to the quinoid moiety of this complex; the closest distance between the
counterion was positioned close to the quinoid moiety of this complex; the closest distance between the  and the C-O carbon atom of the phenoxyl ligand was 3.026 Å. Therefore, one-electron oxidized Pd(II) complexes can be assigned to relatively localized PdII(phenoxyl)(phenolate) complexes.
and the C-O carbon atom of the phenoxyl ligand was 3.026 Å. Therefore, one-electron oxidized Pd(II) complexes can be assigned to relatively localized PdII(phenoxyl)(phenolate) complexes.
The Ni and Pt 5-membered dinitrogen chelate complexes also exhibited a clear symmetrical coordination sphere contraction in both the two M-O and two M-N bond lengths (ca. 0.02 Å) upon oxidation, and the C-O bond distances of these complexes are also shorter than the same bonds before oxidation. These observations suggest that the complexes have the phenoxyl radical characteristics and that the radical electron is delocalized on the two phenolate moieties. Indeed, the XPS and K-edge XANES of an oxidized Ni complex showed the same binding energies and pre-edge peak of the nickel ion as those of the complex before oxidation [42,43,45]. These results supported that the valence state of the nickel ion is +II. On the other hand, NiIII complexes of salen-type ligands can be generated by the axial ligand coordination to the oxidized nickel complex. The X-ray crystal structure of the NiIII-salen-type complex, [Ni (salen)]2O, was reported in 2000 by Mitra et al. (Figure 6 and Table 1) [46]. The NiIII complex was a dinuclear species bridged by an oxo group with a 5-coordinated square pyramidal structure around each NiIII ion. The Nioxo bond is short (1.790(2) Å), while the Ni-phenolate oxygen bonds (1.912 - 1.949 Å) are similar to those of the NiII salen-type complexes. Further the C-O and C-C bond lengths of phenolate moieties in the NiIII complex are also similar to those of the NiII salen-type complexes, suggesting that the oxidation locus of this complex is assigned to the metal center rather than the phenolate moiety. Thus, the high-valent NiIII-phenolate complexes have different structural features from those of the NiII- phenoxyl radical complexes.
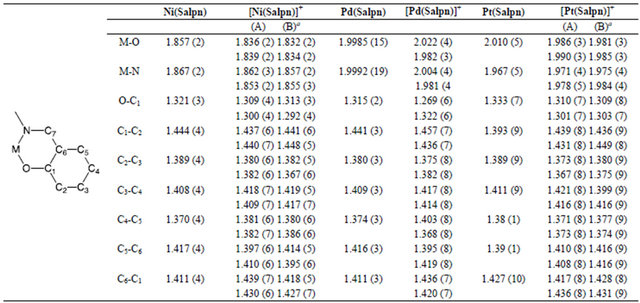
Table 2. Selected bond lengths (Å) of the group 10 metal 6-membered dinitrogen chelate complexes, M(Salpn) and [M(Salpn)]+ complexes.
a(A) and (B) indicates the bond lengths of the two independent molecules (A) and (B) in the unit cell.
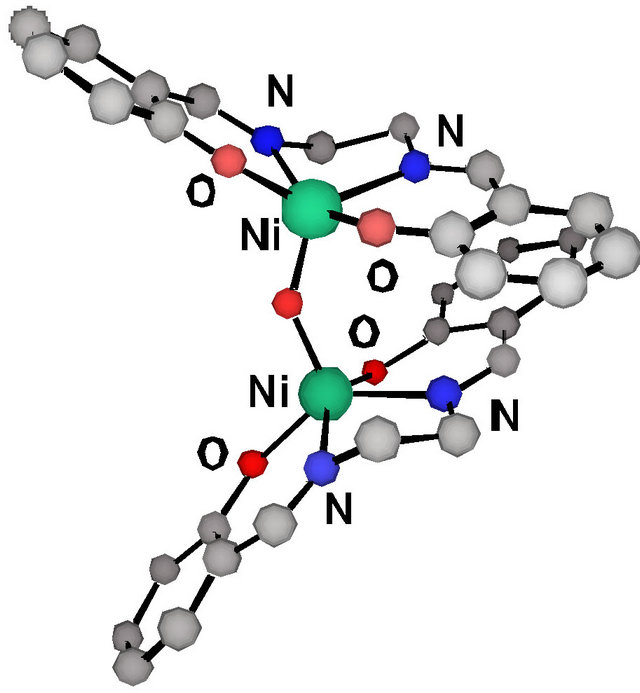
Figure 6. Crystal structure of oxo-bridged dinuclear Ni(III) complex, [Ni(salen)]2O.
However, a close look into the details of the crystal structures reveals that there are subtle differences between them, and especially the oxidized Pd(II) complexes are different from the other complexes [42,43]. Comparison of the 5-membered dinitrogen chelate backbones of the Salcn and Salen complexes indicates that upon oxidation the Ni and Pt complexes exhibited a clear coordination sphere contraction due to shortening of the M-O and M-N bond lengths. On the other hand, the Pd complex showed an unsymmetrical contraction [42]: One of the Pd-O bonds (2.003 Å) is longer than the other (1.963 Å), and the C-O bond (1.263 Å) of the phenolate moiety with the longer Pd-O bond is shorter than the other C-O bond (1.317 Å). The phenolate moiety with a shorter C-O bond length has the lengthening of the ring ortho C-C bonds in comparison with those of the other one. These structural features of the phenolate moiety in the oxidized Pd complex are in good agreement with the characteristics of the phenoxyl radical, which showed the quinoid form due to delocalization of the radical electron on the phenolate moiety as shown in Scheme 2 and reference [53]. Such properties were also detected for the Pd complex with the 6-membered dinitrogen chelate back bone, [Pd(Salpn)]SbF6 [43]. In addition, the 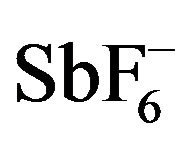 counterion was positioned close to the quinoid moiety of this complex; the closest distance between the
counterion was positioned close to the quinoid moiety of this complex; the closest distance between the 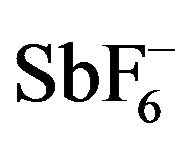 and the C-O carbon atom of the phenoxyl ligand was 3.026 Å. Therefore, one-electron oxidized Pd(II) complexes can be assigned to relatively localized PdII(phenoxyl)(phenolate) complexes.
and the C-O carbon atom of the phenoxyl ligand was 3.026 Å. Therefore, one-electron oxidized Pd(II) complexes can be assigned to relatively localized PdII(phenoxyl)(phenolate) complexes.
In the case of Pt complexes, the XPS of the oxidized complex was slightly different from that before oxidation. The binding energies of the Pt ion in the oxidized complex were +0.2 eV higher, and LIII-edge XANES exhibited an increasing white line. Such spectral features suggest that the oxidation state of the Pt ion in the oxidized complex is higher than +II but that the differences are rather small [45]. From these results, [Pt(Salen)]SbF6 can be described mainly as the Pt(II)-phenoxyl radical species, but the radical electron is fully delocalized over the whole molecule including the central metal ion [43]. Such an electronic structure is similar to that of [Ni (Salcn)]SbF6, and therefore, the X-ray structure of [Pt (Salen)]SbF6 does not show significantly different characteristics from those of [Ni(Salcn)]SbF6.
The six-membered NiII and PtII Salpn chelate complexes are slightly different from the 5-membered dinitrogen chelate Salcn and Salen complexes [44]. The crystal structures of both oxidized Salpn complexes exhibited two crystallographically independent molecules in the unit cell, where the M-O and M-N bond lengths do not differ substantially between the two molecules. The bond lengths in the coordination plane are ca. 0.02 Å shorter than those of the neutral complexes, and this contraction upon oxidation is in good agreement with that in the 5-membered dinitrogen chelate complexes (Table 2). However, the C-O bond lengths of the two phenolate moieties differed for the two independent molecules; one of the molecules showed very similar C-O bond lengths, while the bond lengths in the other molecule were slightly different, showing a similar tendency to that of the oxidized Pd complexes (Table 2). Therefore, the 6-membered chelate Ni and Pt complexes can be considered to be closer to the localized phenoxyl radical metal (II) complexes in comparison with the 5-membered chelate complexes of Salcn and Salen due to the chelate effect of the dinitrogen backbone. Such characteristics of the more localized radical electron in oxidized NiII and PtII Salpn complexes are also observed in the spectroscopies [47], which indicate that the X-ray structures reflect the electronic structures of the complexes.
5.2. Cu(II)-Salen-Type Complexes.
The electronic structure determinations of the Cu complexes are clearly made by X-ray structure analyses. Structures of four one-electron oxidized CuII salen-type complexes are shown in Figure 7 [48-51]. X-ray analyses of all these complexes established that their structures are similar to those before oxidation, indicating a simple one-electron transfer from the precursors. However, these Cu complexes have different electronic structures.
Figure 7 shows that the oxidized Cu(II) complexes have the  counterion at different positions. The crystal structure of the 5-membered chelate dinitrogen backbone complex, [Cu(1,2-Salcn)]SbF6, indicates that a weak axial Cu-F interaction (2.76 Å) exists between the counterion and the metal center [48]. The position of counteranion suggests that [Cu(1,2-Salcn)]SbF6 has a CuIII character. In addition this complex showed contraction of the coordination sphere without shortening of the C-O bonds of both phenolate moieties, and distortion of the coordination plane was substantially reduced from that in Cu(1,2-Salcn). Such structural features are in good agreement with those of the low-spin d8 Cu(III) complexes. Indeed, the XAF and XPS studies of [Cu (1,2-Salcn)]SbF6 revealed the Cu(III)-phenolate ground state [48]. On the other hand, the 6-membered chelate dinitrogen salen-type complex, [Cu(1,3-Salcn)]SbF6, showed a weak interaction between the counterion and one side of the phenolate moieties, suggesting that this complex can be assigned to the CuII-phenoxyl radical [49]. This complex shows that the C-O bond of one of the phenolate moieties is shortened and that the Cu-O bond with the shortened phenolate moiety becomes longer (Table 3). The counterion is close to the phe-
counterion at different positions. The crystal structure of the 5-membered chelate dinitrogen backbone complex, [Cu(1,2-Salcn)]SbF6, indicates that a weak axial Cu-F interaction (2.76 Å) exists between the counterion and the metal center [48]. The position of counteranion suggests that [Cu(1,2-Salcn)]SbF6 has a CuIII character. In addition this complex showed contraction of the coordination sphere without shortening of the C-O bonds of both phenolate moieties, and distortion of the coordination plane was substantially reduced from that in Cu(1,2-Salcn). Such structural features are in good agreement with those of the low-spin d8 Cu(III) complexes. Indeed, the XAF and XPS studies of [Cu (1,2-Salcn)]SbF6 revealed the Cu(III)-phenolate ground state [48]. On the other hand, the 6-membered chelate dinitrogen salen-type complex, [Cu(1,3-Salcn)]SbF6, showed a weak interaction between the counterion and one side of the phenolate moieties, suggesting that this complex can be assigned to the CuII-phenoxyl radical [49]. This complex shows that the C-O bond of one of the phenolate moieties is shortened and that the Cu-O bond with the shortened phenolate moiety becomes longer (Table 3). The counterion is close to the phe-