Journal of Modern Physics
Vol.4 No.3A(2013), Article ID:29365,10 pages DOI:10.4236/jmp.2013.43A062
DFT Study on Reaction Mechanism of DNA Base Pair with Hydroxyl Radical
1Department of Computer Science and Engineering, Toyohashi University of Technology, Toyohashi, Japan
2Department of Molecular and Quantum Biophysics, Institute of Molecular Biology and Genetics, National Academy of Sciences of Ukraine, Kyiv, Ukraine
Email: *kurita@cs.tut.ac.jp, vid@ipnet.kiev.ua
Received December 28, 2012; revised January 30, 2013; accepted February 9, 2013
Keywords: Density Functional Theory; DNA; Hydroxyl Radical; Base Pair; Reaction Mechanism; Dehydrogenation
ABSTRACT
In order to elucidate the indirect effect by radiation on DNA base pairs, we investigate the mechanism for the attacking reaction of a hydroxyl radical (•OH-radical) to the G-C and A-T base pairs, by the density functional theory (DFT) calculations. The effect of solvation on the mechanism is also revealed by performing the same DFT calculations under the continuum solvation approximation. We find the stable structures for the dehydrogenated G-C and A-T base pairs, in which the hydrogen atom of NH2 group of G or A base is abstracted by the •OH-radical. The solvation around the base pairs stabilizes the dehydrogenated structures significantly, indicating the acceleration of the attacking reaction by •OH-radical to the base pairs in water. Therefore, we conclude that the hydrogen atom of the NH2 group of G or A base in the G-C and A-T base pairs is the most preferably abstracted by the •OH-radical in living cells.
1. Introduction
Genetic information recorded in DNA can be damaged by the variety of environmental agents, in particular, such as radiation. There are two types of effects by radiation, which are commonly called direct and indirect effects [1]. Only a small part of high-energy radiation can attack directly to the DNA molecule in living organs to damage its structure and electronic properties [2]. In fact, when a cell is exposed to radiation, the probability of the radiation interacting with the DNA molecule is very small, since it makes up a small part of the cell mostly composed of water molecules. Therefore, there is a much higher probability of radiation interacting with the water molecules. As a result, the principal effect of radiation is the ionization of water molecules in a cell, producing highly reactive hydroxyl radicals (•OH-radical). Because they react with organic cellular components, especially DNA and lipids, DNA is damaged in its structure and electronic properties [3,4]. In solution, the •OH-radicals form hydrogen bonds with solvating water molecules. It is thus expected that the relative orientation of •OH-radical with respect to the hydrogen atoms of DNA bases are restricted, leading to retardation in hydrogen abstraction from the DNA bases by the •OH-radical. Therefore, it is important to investigate the chemical reaction of
•OH-radical to DNA in solvating water molecules for elucidating the initial damage in DNA induced by the attacking of •OH-radicals under a realistic environment in living cells.
There are various effects of the ionizing radiation on the biological systems, ranging from the development of genetic aberrations, carcinogenesis to aging. Most of these effects have attributed to the role of free radicals. In particular •OH-radicals are major contributors in the single/double strand breaking of the DNA duplexes, because about 2/3 of the surrounding environment of the DNA duplex in the cell-nucleus is composed of water molecules [5]. Due to the short (within nano-seconds) life-time of the •OH-radical [6], it is generated only within 1 nm from the surface of the DNA duplex and can remove a hydrogen ion to form a water molecule. The high reactivity of the •OH-radical is attributed to the possible pairing of two electrons having the opposite spin, which come from the •OH-radical and the target molecule. In the solvated environment, •OH-radicals reach an active site of the DNA molecule and remove a hydrogen ion from the sugar moiety (e.g., a deoxyribose) or nucleobase of DNA. As a consequence of the •OH-radical attacking to DNA, the native double-helical structure of DNA is altered leading to errors in the genetic information stored in DNA [7]. Therefore, understanding the damage in DNA induced by the hydrogen abstraction is of particular importance to reveal the effect of •OH-radicals on DNA.
There are a huge number of theoretical studies on the reaction mechanism between DNA and •OH-radicals, based on classical Monte Carlo (MC) [8], classical molecular dynamics (MD) [9] or quantum mechanical density functional theory (DFT) [10-16] method. It is indispensable to perform quantum mechanical molecular orbital (MO) or DFT calculation for the complex of DNA with •OH-radical, in order to elucidate the reaction mechanism at an electronic level. Since guanine is the most likely to be attacked by •OH-radicals among the four DNA nucleobases [5], many quantum mechanical simulations were performed for guanine. For example, the gas-phase dehydrogenation reaction of guanine with •OH-radical was studied by Car-Parinello first-principle MD simulations [10], while the DFT calculations [11,12] were carried out for the complex of guanine with •OHradical. In addition, the •OH-radical attacking reaction to G-C base pair was investigated by DFT calculations [13, 14]. On the other hand, a limited number of MO or DFT calculations were performed for the adenine base [15] and A-T base pair [16]. And to our knowledge, there is no MO or DFT calculation investigating the difference in the dehydrogenation reaction between G-C and A-T base pairs induced by the •OH-radical.
In the present study, in order to elucidate the indirect effect by radiation on DNA base pairs, we investigate the stable structures and their electronic properties for the complexes of G-C or A-T base pair with an •OH-radical, by the DFT calculations in vacuum and in water. The results reveal that the •OH-radical prefers to react with the NH2 group of G (A) base of G-C (A-T) base pair and to abstract the hydrogen atom of the NH2 group, resulting in a water molecule. These abstractions can produce the isomerization of G and A bases, leading to spontaneous mutations in DNA base sequence.
2. Details of DFT Calculations
Since DFT method includes electron correlation effect and describes the electronic properties of molecules and solids enough accurately, we used this method in the present study. We first constructed the structures of Watson-Crick type conventional G-C and A-T base pairs and optimized their structures in vacuum. For this optimization, we employed the DFT program D-mol3 [17,18], and the Kohn-Sham orbitals were expanded in a numerical basis set DNP (double-numerical plus d-polarization functions for heavy atoms and p-polarization functions for hydrogen atom). The revised Perdew-Burke-Ernzerhof (RPBE) functional [19] of generalized gradient approximation was employed for exchange and correlation potentials. In addition, to confirm the accuracy of the above DFT calculation, the G-C and A-T structures were optimized by the B3LYP [20] hybrid density functional in conjunction with the 6-31G(d,p) basis-set using the Gaussian 09 suite of program [21]. The previous DFT calculation [22] confirmed that the B3LYP functional can describe the potential energy surface for the reaction mechanisms of •OH-radical with imidazole, tyrosine, pyrimidine and purine. In the present DFT study, the terminal sites connecting to the C1’ atom of deoxyribose in DNA strands were terminated by the CH3 group, as shown in Figure 1.
In order to find the most preferable attacking site of •OH-radical around the G-C and A-T base pairs, we added an •OH-radical near the hydrogen atom of the base pair and constructed the six types of initial structures for the base pair + •OH-radical complexes, as shown in Figures 2 and 3. The distance between the hydrogen atom of base and the oxygen atom of •OH-radical was set as 1.5 Å. These structures were optimized in vacuum by the unrestricted DFT method (RPBE/DNP) of D-mol3, with a doublet spin state assigned. In addition, to investigate the effect of solvation on the reaction mechanism between the base pair and the •OH-radical, we performed the same DFT optimizations in water approximated by the continuum solvation model [23] of D-mol3.
3. Results and Discussion
3.1. Optimized Structures of DNA Base Pair + •OH Radical in Vacuum We first optimized the G-C and A-T structures in vacuum

Figure 1. Optimized structures of base pairs: (a) G-C and (b) A-T.
 (a)
(a) (b)
(b) (c)
(c) (d)
(d) (e)
(e) (f)
(f)
Figure 2. Initial structures of G-C base pair + •OH-radical; (a) Case 1; (b) Case 2; (c) Case 3; (d) Case 4; (e) Case 5; and (f) Case 6.
 (a)
(a) (b)
(b) (c)
(c) (d)
(d) (e)
(e) (f)
(f)
Figure 3. Initial structures of A-T base pair + •OH-radical; (a) Case 1; (b) Case 2; (c) Case 3; (d) Case 4; (e) Case 5; and (f) Case 6.
by the DFT method (RPBE/DNP). The optimized structures are shown in Figure 1. The obtained hydrogen bond lengths between G and C and between A and T bases are identical within a 0.02 Å difference to those for the structures optimized by the present B3LYP/6-31G(d,p) calculations, the previous B3LYP/6-31G(d) calculations [16] as well as those in the B-form DNA structure obtained by the X-ray analysis [24]. Accordingly, the present DFT calculation is confirmed to be valid for obtaining the stable structures of the closed-shell base-pairs.
To elucidate which hydrogen atom of G-C and A-T base pairs can be abstracted by the •OH-radical, we added it near the hydrogen atom of the base pairs to construct the six types of initial structures shown in Figures 2 and 3. They were fully optimized by the unrestricted DFT calculations, with a doublet spin state assigned. Figures 4 and 5 show the structures optimized in vacuum. As in the previous DFT calculations [16], the •OH-radical can be stabilized near the hydrogen bond between the DNA bases. In addition, we found a stable structure shown in Figure 4(d), in which the •OH-radical abstracts a hydrogen atom from the NH2 group of guanine in G-C base pair to change into H2O. This structure is at least 10.6 kcal/mol more stable than the other optimized structures, as listed in Table 1(a). In contrast, the other hydrogen atoms of G-C base pair can not be abstracted by the •OH-radical, as indicated in Figure 4. Therefore, we can conclude that the •OH-radical prefers to react with the NH2 group of guanine of G-C base pair in vacuum and abstract the hydrogen atom of the NH2 group, as indicated in Figure 4(d).
This result for G-C is comparable to the recent experimental results [25] obtained by absorption spectroscopy, which imply that the major reaction between guanine and the •OH-radical is hydrogen abstraction from the NH2 group. In contrast, the other experiments [26,27] using the redox titration technique indicate that the •OH-radical adds to the C4, C5 or C8 positions on purine bases, leading

Figure 4. Optimized structures of G-C base pair + •OH-radical in vacuum; (a) Case 1; (b) Case 2; (c) Case 3; (d) Case 4; (e) Case 5; and (f) Case 6. Total energy (kcal/mol) relative to the most stable structure and the distance (Å) between the oxygen atom of •OH and the hydrogen atom of DNA base are shown.

Figure 5. Optimized structures of A-T base pair + •OH-radical in vacuum; (a) Case 1; (b) Case 2; (c) Case 3; (d) Case 4; (e) Case 5; and (f) Case 6. Total energy (kcal/mol) relative to the most stable structure and the distance (Å) between the oxygen atom of •OH and the hydrogen atom of DNA base are shown.
Table 1. Total energies (T.E.) (kcal/mol) and their relative energies (ΔT.E.) to the most stable structure for the optimized structures of the G-C/A-T + •OH-radical complexes shown in Figures 4-7 obtained by RPBE/DNP. (a) In vacuum; (b) In water.

to ring-opening or dehydration reaction. As for the molecular simulation on the reaction between G-C and •OH-radical, state-of-the-art ONIOM calculations based on DFT were performed [13] for an elaborated hydrated DNA model. The computed results demonstrate that the attack of •OH-radical on the cytosine base increases the strength of O6(G)-N4(C) bond and consequently the efficiency of the proton transfer reactions. However, there is no result about the abstraction of the hydrogen atom from G-C base pair induced by the •OH-radical.
Figure 5 shows the optimized structures for the complex of A-T base pair with •OH-radical, indicating that the hydrogen atoms of A-T can not be extracted by •OHradical in vacuum. Among the six structures optimized in the present study, the structure shown in Figure 5(c) is the most stable. Even in the most stable structure, the •OH-radical can not abstract the hydrogen atom from the NH2 group of adenine base, although the •OH-radical is stabilized near the NH2 group. Consequently, it seems that A-T base pair is relatively insulated from the influence of •OH-radicals in vacuum.
3.2. Optimized Structures of DNA Base Pair + •OH-Radical in Water
Since DNA is surrounded by a large number of water molecules in a cell, their effect should be considered in the analysis for the •OH-radical attacking reaction mechanism. In principle, the water molecules should be considered explicitly in the DFT calculations. However, in the practice of the DFT calculations, it is very timeconsuming task to perform the DFT optimizations for the fully-solvated systems composed of DNA base pair and •OH-radical. We thus considered the effect of solvating water molecules by the continuum solvation model [23] as the first step of the DFT simulations. The initial structures of G-C and A-T base pairs with •OH-radical shown in Figures 2 and 3 were fully optimized in water approximated by the continuum solvation model. Figures 6 and 7 show these optimized structures, and their total

Figure 6. Optimized structures of G-C base pair + •OH-radical in water; (a) Case 1; (b) Case 2; (c) Case 3; (d) Case 4; (e) Case 5; and (f) Case 6. Total energy (kcal/mol) relative to the most stable structure and the distance (Å) between the oxygen atom of •OH and the hydrogen atom of DNA base are shown.

Figure 7. Optimized structures of A-T base pair + •OH-radical in water; (a) Case 1; (b) Case 2; (c) Case 3; (d) Case 4; (e) Case 5; and (f) Case 6. Total energy (kcal/mol) relative to the most stable structure and the distance (Å) between the oxygen atom of •OH and the hydrogen atom of DNA base are shown.
energies are listed in Table 1(b).
As shown in Figures 6(a) and (d), for the G-C base pair, the two stable structures with the hydrogen abstraction from the NH2 group of guanine were obtained in water. They are at least 15.6 kcal/mol more stable than the other structures shown in Figure 6. Therefore, it is concluded that the abstraction of the hydrogen atom from the NH2 group of guanine is energetically more favorable in water. If •OH-radical can come close to the guanine NH2 group of the DNA duplex in a cell, the •OH-radical is expected to abstract the hydrogen atom from the guanine base and cause damage to the base. To elucidate the reaction mechanism between G-C and •OH-radical, the ab initio MD simulations based on DFT are underway now in the continuum salvation model. The results will be published elsewhere.
The effect of solvation is also significant for the reaction of •OH-radical to A-T base pair. As shown in Figure 7(c), •OH-radical can attack and abstract the hydrogen atom of the adenine NH2 group of A-T base pair in water. This structure is at least 15.5 kcal/mol more stable than the other structures shown in Figure 7, indicating that the dehydrogenated A-T structure shown in Figure 7(c) is largely preferable in water. As mentioned above, the present DFT optimizations in water elucidate that the hydrogen atom of the NH2 group of guanine and adenine bases in the solvated G-C and A-T base pairs can be abstracted by the •OH-radical from the view points of energy.
As for the •OH-radical reaction with adenine, the energetics and associated transition states for the dehydrogenation of adenine by •OH-radical were investigated by DFT calculations [15]. The results elucidated that the reaction is exothermic and that the N6 position is most favorable for the •OH-radical attack, resulting in the formation of N6-dehydrogenated adenine radicals. This result is consistent with our present results on A-T base pair obtained in water. Consequently, it can be concluded that the hydrogen atom of the NH2 group of adenine of A-T prefers to be abstracted by the •OH-radical attack.
In order to elucidate the reason why the hydrogen atom of NH2 group of guanine and adenine bases in the G-C and A-T base pairs is abstracted by the •OH-radical attack, we investigated the electronic properties of these base pairs in vacuum and in water by the DFT calculations. Figures 8 and 9 show the space distributions of highest occupied MO (HOMO) and lowest unoccupied MO (LUMO) for G-C and A-T, respectively. Since the HOMO levels of these base pairs are higher in energy than the singly occupied MO (SOMO) level of •OHradical, it is expected that the •OH-radical reacts with the base pairs at the position with larger distribution of HOMO. As shown in Figures 8(a) and (c), the HOMO of G-T is distributed on guanine both in vacuum and in water. Among the hydrogen atoms of guanine, those of NH2 group and that of CH group have larger HOMO distribution. The CH bond is stronger than the NH bonds of NH2 group. As a result, the •OH-radical prefers to react with the hydrogen atom of the NH2 group and abstract it in the G-C base pair. As indicated in Figure 9, the
 (a)
(a) (b)
(b) (c)
(c) (d)
(d)
Figure 8. Space distributions of HOMO and LUMO for G-C base pair in vacuum and in water. (a) G-C HOMO in vacuum; (b) G-C LUMO in vacuum; (c) G-C HOMO in water; (d) G-C LUMO in water.
 (a)
(a) (b)
(b) (c)
(c)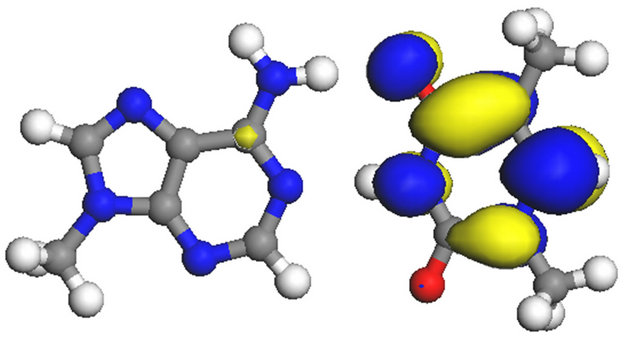 (d)
(d)
Figure 9. Space distributions of HOMO and LUMO for A-T base pair in vacuum and in water. (a) A-T HOMO in vacuum; (b) A-T LUMO in vacuum; (c) A-T HOMO in water; (d) A-T LUMO in water.
situation is similar for the A-T base pair. HOMO is distributed on adenine, and the NH2 group of adenine has significant HOMO distribution. These HOMO distributions for G-C and A-T base pairs can be related with the formation of the dehydrogenated structures of the NH2 group of guanine and adenine induced by •OH-radical.
4. Conclusions
By using the DFT calculations, we investigated the stable structures for the complexes of G-C and A-T base pairs with •OH-radical in vacuum and in water, which was described by the continuum solvation model.
The results are summarized as follows:
1) In vacuum, the hydrogen atom of NH2 group of guanine base in G-C base pair is abstracted by the •OHradical, while that of adenine base is not abstracted.
2) In continuum solvation model, the •OH-radical abstracts the hydrogen atom of the NH2 group of G and A bases in both the G-C and A-T base pairs.
3) The reaction of •OH-radical attack to G-C or A-T base pair is exothermic and accelerated by the effect of solvation.
5. Acknowledgements
This work was supported in part by grants from the JSPS Grant-in-Aid for Challenging Exploratory Research (No. 22650061), the Murata Science Foundation, the Iketani Science and Technology Foundation, the Tatematsu Foundation and the CASIO Science Promotion Foundation.
REFERENCES
- T. Douki and J. Cadet, “Radiation-Induced Damage to DNA: From Model Compounds to Cell,” In: M. Spotheim-Maurizot, M. Mostafavi, T. Douki and J. Belloni, Eds., Radiation Chemistry: From Basics to Applications in Material and Life Sciences, EDP Sciences, Les Ulis Cedex A, 2008, pp. 177-189.
- D. Khanduri, S. Collins, A. Kumar, A. Adhikary and M. D. Sevilla, “Formation of Sugar Radicals in RNA Model Systems and Oligomers via Excitation of Guanine Cation Radical,” Journal of Physical Chemistry B, Vol. 112, No. 7, 2008, pp. 2168-2178. doi:10.1021/jp077429y
- J. Cadet, T. Douki and J.-L. Ravanat, “Oxidatively Generated Base Damage to Cellular DNA,” Free Radical Biological Medicine, Vol. 49, No. 1, 2010, pp. 9-21. doi:10.1016/j.freeradbiomed.2010.03.025
- J. R. Wagner and J. Cadet, “Oxidation Reactions of Cytosine DNA Components by Hydroxyl Radical and OneElectron Oxidants in Aerated Aqueous Solutions,” Accounts of Chemical Research, Vol. 43, No. 4, 2010, pp. 564-571. doi:10.1021/ar9002637
- B. Aydogan, W. E. Bolch, S. G. Swarts, J. E. Turner and D. T. Marshall, “Monte Carlo Simulations of Site-Specific Radical Attack to DNA Bases,” Radiation Research, Vol. 169, No. 2, 2008, pp. 223-231. doi:10.1667/RR0293.1
- H. Sies, “Strategies of Antioxidant Defense,” European Journal of Biochemistry, Vol. 215, No. 2, 1993, pp. 213- 219. doi:10.1111/j.1432-1033.1993.tb18025.x
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunce, “Oxidative DNA Damage: Mechanisms, Mutation, and Disease,” FASEB Journal, Vol. 17, No. 10, 2003, pp. 1195- 1214. doi:10.1096/fj.02-0752rev
- H. Tomita, M. Kai, T. Kusama and A. Ito, “Monte Carlo Simulation of DNA Strand-break Induction in Supercoiled Plasmid pBR322 DNA from Indirect Effects,” Radiation and Environmental Biophysics, Vol. 36, No. 4, 1998, pp. 235-241. doi:10.1007/s004110050077
- R. M. Abolfath, A. C. T. van Duin and T. Brabec, “Reactive Molecular Dynamics Study on the First Steps of DNA Damage by Free Hydroxyl Radicals,” Journal of Physical Chemistry A, Vol. 115, No. 40, 2011, pp. 11045- 11049. doi:10.1021/jp204894m
- C. J. Mundy, M. E. Colvin and A. A. Quong, “Irradiated Guanine: A Car-Parrinello Molecular Dynamics Study of Dehydrogenation in the Presence of an OH Radical,” Journal of Physical Chemistry A, Vol. 106, No. 43, 2002, pp. 10063-10071. doi:10.1021/jp0212904
- A. Kumar, V. Pottiboyina and M. D. Sevilla, “Hydroxyl Radical (OH) Reaction with Guanine in an Aqueous Environment: A DFT study,” Journal of Physical Chemistry B, Vol. 115, No. 50, 2011, pp. 15129-15137. doi:10.1021/jp208841q
- R. M. Abolfath, P. K. Biswas, R. Rajnarayanam, T. Brabec, R. Kodym and L. Papiez, “Multiscale QM/MM Molecular Dynamics Study on the First Steps of Guaninedamage by Free Hydroxyl Radicals in Solution,” Journal of Physical Chemistry A, Vol. 116, No. 15, 2012, pp. 3940- 3945. doi:10.1021/jp300258n
- J. P. Ceron-Carrasco and D. Jacquemin, “Interplay between Hydroxyl Radical Attack and H-bond Stability in Guanine-Cytosine,” RSC Advances, Vol. 2, No. 31, 2012, pp. 11867-11875. doi:10.1039/c2ra22389a
- R. B. Zhang and L. A. Eriksson, “Effects of OH Radical Addition on Proton Transfer in the Guanine-Cytosine Base Pair,” Journal of Physical Chemistry B, Vol. 111, No. 23, 2007, pp. 6571-6576. doi:10.1021/jp071772l
- Q. Cheng, J. Gu, K. R. Compaan and H. F. Schaefer III, “Hydroxyl Radical Reactions with Adenine: Reactant Complexes, Transition States, and Product Complexes,” Chemistry—A European Journal, Vol. 16, No. 39, 2010, pp. 11848-11858. doi:10.1002/chem.201001236
- Y. Maruyama, M. Tachikawa and S. Kawano, “Ab Initio Study of DNA Double-Strand Breaks by Hydroxyl Radicals,” Japanese Society Mechanism Enginering International Journal B, Vol. 48, 2005, pp. 196-201.
- B. Delley, “An All Electron Numerical Method for Solving the Local Density Functional for Polyatomic Molecules,” Journal of Chemical Physics, Vol. 92, No. 1, 1990, pp. 508-517. doi:10.1063/1.458452
- B. Delley, “From Molecules to Solids with the DMol3 Approach,” Journal of Chemical Physics, Vol. 113, No. 18, 2000, pp. 7756-7764. doi:10.1063/1.1316015
- J. Perdew, K. Burke and M. Ernzerhof, “Generalized Gadient Aproximation Made Simple,” Physical Review Letters, Vol. 77, 1996, pp. 3865-3868. doi:10.1103/PhysRevLett.77.3865
- A. D. Becke, “Density-Functional Thermochemistry. III. The Role of Exact Exchange,” Journal of Chemical Physics, Vol. 98, No. 7, 1993, pp. 5648-5652. doi:10.1063/1.464913
- M. J. Frisch, et al., “Gaussian 09,” Gaussian, Inc., Wallingford, 2009.
- M. J. Lundqvist and L. A. Eriksson, “Hydroxyl Radical Reactions with Phenol as a Model for Generation of Biologically Reactive Tyrosyl Radicals,” Journal of Physical Chemistry B, Vol. 104, No. 4, 2000, pp. 848-855. doi:10.1021/jp993011r
- B. Delley, “The Conductor-like Screening Model for Polymers and Surfaces,” Molecular Simulation, Vol. 32, No. 2, 2006, pp. 117-123. doi:10.1080/08927020600589684
- S. Arnott and D. W. L. Hukins, “Refinement of the Structure of B-DNA and Implication for the Analysis of the X-Ray Diffraction Data from Fibers of Biopolymers,” Journal of Molecular Biology, Vol. 81, No. 2, 1973, pp. 93-105. doi:10.1016/0022-2836(73)90182-4
- C. Chatgilialoglu, M. D’Angelantonio, M. Guerra, P. Kaloudis and Q. G. Mulazzani, “A Reevaluation of the Ambident Reactivity of the Guanine Moiety Towards Hydroxyl Radicals,” Angewandte Chemie International Edition, Vol. 48, No. 12, 2009, pp. 2214-2217. doi:10.1002/anie.200805372
- S. Steenken, “Purine Bases, Nucleosides, and Nucleotides: Aqueous Solution Redox Chemistry and Transformation Reactions of Their Radical Cations and e− and OH Adducts,” Chemical Reviews, Vol. 89, No. 3, 1989, pp. 503- 520. doi:10.1021/cr00093a003
- A. J. S. C. Vieira and S. Steenken, “Pattern of Hydroxy Radical Reaction with Adenine and Its Nucleosides and Nucleotides. Characterization of Two Types of Isomeric Hydroxy Adduct and Their Unimolecular Transformation Reactions,” Journal of the American Chemical Society, Vol. 112, No. 19, 1990, pp. 6986-6994. doi:10.1021/ja00175a036
NOTES
*Corresponding author.