Journal of Environmental Protection
Vol.05 No.12(2014), Article ID:49998,18 pages
10.4236/jep.2014.512111
Extraction Characteristics of Selenium as Affected by Coal Fly Ash Type, Water Extractant, and Extraction Time
Mark A. Cantrell1, Kristofor R. Brye2, David M. Miller2, Esten Mason2, Julian Fairey3
1AEP-Swepco, Flint Creek Power Plant, Gentry, AR, USA
2Department of Crop, Soil, and Environmental Sciences, University of Arkansas, Fayetteville, AR, USA
3Department of Civil Engineering, University of Arkansas, Fayetteville, AR, USA
Email: kbrye@uark.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 14 July 2014; revised 12 August 2014; accepted 5 September 2014
ABSTRACT
Selenium (Se) contamination can be a potential groundwater concern near un-lined coal ash landfills. Of all the Environmental Protection Agency’s priority and non-priority pollutants, Se has the narrowest concentration range considered beneficial and detrimental for aquatic and terrestrial organisms. The effects of ash type (i.e., fresh and weathered), water-extractant type (i.e., deionized water, rainwater, and groundwater), and extraction time (i.e., 2 and 6 hours) on Se, arsenic (As), and chromium (Cr) concentrations were investigated from Class C, subbituminous coal fly ash produced at the Flint Creek Power Plant (Benton County, AR). Water-extractable Se concentrations differed (p = 0.03) between ash types across water-extractants, but were unaffected (p > 0.05) by extraction times. Unexpectedly, fresh ash water-extractable Se concentrations were below minimum detection limits (i.e., 2.0 µg∙L−1) for all treatments. In contrast, averaged over extraction times, the water-extractable Se concentration from weathered ash was greatest (p < 0.05) with groundwater and rainwater, which did not differ and averaged 60.0 µg∙L−1, compared to extraction with deionized water (57.6 µg∙L−1). Selenite 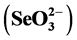 was greater (p < 0.001) in the fresh (3.85 mg∙kg−1) than in the weathered ash (0.70 mg∙kg−1), while selenate
was greater (p < 0.001) in the fresh (3.85 mg∙kg−1) than in the weathered ash (0.70 mg∙kg−1), while selenate 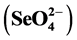 concentration was greater (p < 0.001) in the weathered (0.67 mg∙kg−1) than in fresh ash (0.48 mg∙kg−1). Results from this study indicate that environmental weathering of Class C, subbituminous fly ash promotes oxidation of selenite, to the less toxic, but highly mobile selenate. The formation of hydrated ettringite [Ca6Al2(SO4)3(OH)12∙26H2O] and calcium selenite (CaSeO3) likely acted as a sink for weathered ash selenite. Implications of this research include a better understanding of the past, present, and future environmental and health risk potential associated with the release of water- soluble Se, As, and Cr to aid in the development of sustainable fly ash management strategies.
concentration was greater (p < 0.001) in the weathered (0.67 mg∙kg−1) than in fresh ash (0.48 mg∙kg−1). Results from this study indicate that environmental weathering of Class C, subbituminous fly ash promotes oxidation of selenite, to the less toxic, but highly mobile selenate. The formation of hydrated ettringite [Ca6Al2(SO4)3(OH)12∙26H2O] and calcium selenite (CaSeO3) likely acted as a sink for weathered ash selenite. Implications of this research include a better understanding of the past, present, and future environmental and health risk potential associated with the release of water- soluble Se, As, and Cr to aid in the development of sustainable fly ash management strategies.
Keywords:
Arsenic, Chromium, Coal Fly Ash, Landfill, Selenium

1. Introduction
In the United States (US), coal-fired plants provide approximately 45% of the total net electrical generation, with 847,930 metric tons of coal consumed per day [1] . During the combustion of coal, inorganic constituents that are naturally enriched in the coal are further concentrated in the coal combustion residuals (CCRs): bottom ash and fly ash. Bottom ash is a coarse, heavy ash that is too dense to carry over in the gas stream after combustion [2] [3] . Due to its density, bottom ash is collected at the bottom of the boiler. The particle size ranges from gravel to silt, with much of the bottom ash sized similar to that of natural sand [3] . Some beneficial re-use options of bottom ash include construction fill material, snow and ice control, concrete/cement products, mining applications, and as road and sub-surface material [3] [4] . The remaining unmarketable bottom ash is either land-filled or placed in impoundments or settling ponds for temporary storage.
The physical and chemical properties of fly ash are largely dependent on the mineral content, composition, and source of the coal, and on the combustion conditions within the boiler [5] . The four major constituents of fly ash are silica (SiO2), aluminum oxide (Al2O3), calcium oxide (CaO), and iron oxide (Fe2O3) [6] [7] . The minor constituents present are magnesium oxide (MgO), sodium oxide (Na2O), titanium oxide (TiO2), potassium oxide (K2O), phosphorus oxide (P2O3), and sulfur trioxide (SO3), the sum of which account for less than 5% of the total weight [6] [7] . There are also trace amounts of heavy metals, such as arsenic, barium, cadmium, chromium, copper, lead, mercury, molybdenum, nickel, selenium, strontium, vanadium, and zinc, which typically do not ex- ceed 1% by weight [8] . Fly ash particles range in size from 0.5 (clay) to 100 µm (fine sand), with a specific gravity that ranges from 2.2 to 2.8 [2] [8] .
According to ASTM [9] , fly ash is classified as Class C or Class F. Class C fly ash generally has more than 20% CaO, more than 50% combined SiO2, Al2O3, and Fe2O3 content, and is normally produced from sub-bituminous or lignite coal combustion. Class F fly ash is usually produced from bituminous and anthracite coal combustion and has less than 10% CaO content and more than 70% SiO2, Al2O3, and Fe2O3 content [9] . Coal samples tested during research conducted by EPRI [6] showed Class C fly ashes having a 14.6% to 27.2% CaO composition and Class F having a 0.3% to 6.8% CaO composition. Approximately 40% of the fly ash produced is re-used in industrial applications, with the remainder being land-filled [10] . Fly ash is typically alkaline and has a high calcium (~27%) content and exhibits pozzolanic properties, much like that of Portland cement [7] [11] [12] . These characteristics allow fly ash to be sold and utilized as an additive in concrete, used as a soil amendment to improve soil structure and water-holding capacity, act as a neutralizer in acidic soils, and serve as a source of essential micronutrients for agricultural crops under the right conditions [7] [8] [11] [13] [14] .
Of the inorganic constituents concentrated in the CCRs, especially fly ash, selenium (Se) is of particular concern due to the transference of Se from the coal to the ash through a physical, volatilization-condensation adsorption process [15] . Fly ash has a mean Se concentration of 14.0 mg Se kg−1 and typically ranges between 5.5 and 46.9 mg Se kg−1 [16] . Out of all of the Environmental Protection Agency’s (EPA) priority and non-priority pollutants, Se has the narrowest concentration range between what is considered beneficial and detrimental to aquatic species and terrestrial organisms. In aquatic ecosystems, water concentrations greater than 5 µg Se L−1 can potentially cause teratogenic deformities within fish populations and embryo toxicity of waterfowl through the bioaccumulative effects of Se in the aquatic food chain [17] - [19] . Humans have a narrow exposure range between what is considered deficient (<40 μg Se day−1) and what is considered toxic (>400 μg Se day−1) [20] . Therefore, the EPA’s maximum contaminant level (MCL) in drinking water for humans is 50 μg Se L−1 [21] .
Since approximately 43,900,000 metric tons of bottom ash and fly ash are stored in coal ash landfills or impoundments in the US annually [10] , leachate generated from a coal ash landfill due to environmental exposure can be laden with Se, and could cause substantial harm if released into the environment. Through environmental weathering, Se can be released from stored fly ash and become mobilized in groundwater if the landfill does not have a properly engineered liner [22] - [24] . The Se-laden leachate can be transported with the groundwater and become a human and ecological threat. Consequently, a better understanding of Se’s mobile, water-soluble fractions is needed.
The water-soluble fraction is the readily accessible fraction that can be easily released into the environment and mobilized through contact with rainwater and groundwater, and therefore is the fraction of greatest environmental concern [25] [26] . Once released into the environment, Se usually exists as the highly mobile selenate 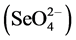 or as the more toxic, but far less mobile selenite
or as the more toxic, but far less mobile selenite 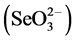 [27] [28] . Changes in oxidation states affect Se’s solubility, (re)adsorption, and mobility rates in soils [29] - [31] . Selenite behaves similar to phosphate (i.e., forms an inner-sphere surface complex), has a greater adsorption rate than selenate, and is controlled by its ability to bind to ferric hydroxides and aluminum oxides in acidic and neutral soils [32] - [35] . In contrast, selenate is known to behave like the sulfate anion (i.e., forms an outer-sphere surface complex) with low adsorption rates, high solubility, and pronounced leachability causing selenate to be highly mobile in soils [29] [36] [37] . Recognizing and understanding the differences between species are critical for assessing the amount of Se that has been, and could be, released into the environment from an ash landfill and to better predict Se’s natural mitigation, such as biotic or abiotic reduction of selenate to selenite and its subsequent precipitation and/or adsorption of selenite to Fe/Al oxides in soils.
[27] [28] . Changes in oxidation states affect Se’s solubility, (re)adsorption, and mobility rates in soils [29] - [31] . Selenite behaves similar to phosphate (i.e., forms an inner-sphere surface complex), has a greater adsorption rate than selenate, and is controlled by its ability to bind to ferric hydroxides and aluminum oxides in acidic and neutral soils [32] - [35] . In contrast, selenate is known to behave like the sulfate anion (i.e., forms an outer-sphere surface complex) with low adsorption rates, high solubility, and pronounced leachability causing selenate to be highly mobile in soils [29] [36] [37] . Recognizing and understanding the differences between species are critical for assessing the amount of Se that has been, and could be, released into the environment from an ash landfill and to better predict Se’s natural mitigation, such as biotic or abiotic reduction of selenate to selenite and its subsequent precipitation and/or adsorption of selenite to Fe/Al oxides in soils.
Considering that relatively little is known about fly-ash-derived Se behavior in the environment, the objective of this study was to evaluate the effects of ash type (i.e., fresh and weathered), water-extractant type (i.e., deionized water, rainwater, and groundwater), and extraction time (i.e., 2 and 6 hours) on water-extractable Se, arsenic (As), and chromium (Cr) concentrations from a Class C, sub-bituminous fly ash. It was hypothesized that water-extractable Se concentrations would be greatest from weathered ash extracted with deionized water, with little to no difference between extraction times, compared to fresh. It was also hypothesized that the dominant Se species would differ between ash types. Comparison of the three different water extractants will provide the scientific community with useful information to help make an informed decision on a preferred water extractant for Se, As, and/or Cr.
2. Materials and Methods
2.1. Ash Production and Disposal
The ash used in this study was obtained from the Flint Creek Power Plant in Benton County, Arkansas (N 36˚15'45" latitude and W 94˚31'15" longitude). The Flint Creek Power Plant is a base-load, coal-fired, electric generation plant that went operational in 1978. The land surrounding the plant is largely rural. The Flint Creek Power Plant was the first commercial power plant in Arkansas to use coal as its fuel source (C. Gregory, Energy Production Supt. II, personal communication, 2010). The plant has the electrical capacity to produce 528 net megawatts per hour, which is enough electricity to supply approximately 365,000 households at an average Arkansas household usage of 1076 kW per month [38] .
At full capacity, the plant will burn approximately 6350 metric tons of coal per day [39] . The Flint Creek Power Plant uses Powder River Basin (PRB) coal mined from the Wyodak Beds in Wyoming. This PRB coal is a Class C, sub-bituminous coal with a heat value of 19,306 to 26,749 kJ∙kg−1 (8,300 to 11,500 Btu∙1b −1) and is one of the cleanest-burning coals available in the US with respect to sulfur dioxide emissions [40] [41] . A typical analysis of sub-bituminous coal (Table 1) yields the following mean elemental content: carbon (69.7%), hydrogen (4.8%), oxygen (17.9%), nitrogen (0.9%), and sulfur (0.3%). The ash produced from burning sub-bituminous coal produces an alkaline, calcium-based ash different from the acidic ash produced from burning eastern US bituminous coals. The coal being used at Flint Creek has a total ash content ranging from 3% to 5%. Before being injected into the boiler, the coal is pulverized into a fine talcum-powder-like consistency. The pulverized coal is then injected into the boiler with pre-heated, forced-draft air. The combustion of the coal produces temperatures over 1370˚C inside the boiler, eventually producing high-pressure steam delivered to the turbine at 169 kg∙cm−2 and 538˚C [39] .
Of the ash produced at Flint Creek, approximately 70% (9.3 metric tons∙hr−1) is fly ash and 30% is bottom ash. Flint Creek uses PRB sub-bituminous coal, which produces a Class C fly ash during combustion. The color of the fly ash produced varies from a yellowish to light tan. Fly ash is transported out of the boiler in the gas stream, and is then collected by the electrostatic precipitator (ESP) and silo baghouse.
Flint Creek uses hot-side ESPs that receive the flue gas at a maximum temperature of 399˚C with a dust loading of 2.0 to 2.5 grains∙m−3 [39] . Flint Creek ESPs use anodic plates and cathodic wires to remove fly ash from the flue gas with greater than 99.5% removal efficiency [42] [43] . Electrostatic precipitation, in this case, is the

Table 1. Analysis of sub-bituminous coal used at the Flint Creek Power Plant for electrical generation [71] .
aSamples were collected and analyzed at the Rochelle/North Antelope mine (Wyodak-Anderson Seam) in Wyoming.
process in which fly ash suspended in the flue gas is given a negative charge from negatively charged electrodes (cathodic wires) hanging in the ESP. The negatively charged fly ash is then collected onto the positively charged (anodic) plates [42] . The plates are periodically vibrated causing the fly ash to fall and be collected in hoppers located below the ESP. Fly ash collected in the silo is either sold or stored on-site in a 16-ha ash landfill located approximately 400 m northeast of the power plant [44] .
Only Class 3 waste generated at the Flint Creek Power Plant is disposed of in the on-site ash landfill. Class 3 waste includes CCRs such as fly ash, bottom ash, bottom ash dredged from settling ponds, slag, and grit-blasting materials. Slag is a glassy, angular, non-crystalline material that accumulates in the boiler as the coal ash melts into a viscous liquid and is quenched for removal. Grit-blasting material is a non-hazardous substance used to remove ash that adheres to the interior surfaces of the boiler, ESPs, and duct work during plant outages [43] .
2.2. Ash Acquisition and Characterization
Grab samples of fly ash were collected at the Flint Creek plant using an on-site InterSystems Model XE0118 automated collection device (InterSystems, Omaha, Ne) installed on the fly ash collection piping on the outlet of the hot-side ESP, referred to hereafter as fresh ash. The ash collector secures approximately 5 g of ash, every 10 minutes, for a total of 30 days during normal plant operation. Fly ash was also collected randomly from the ash landfill at a depth of approximately 1.0 m using a backhoe, hereafter referred to as weathered ash. The actual age of the weathered ash was unknown due to the historic disposal methods of the ash. However, weathered samples were collected from an area of the landfill that was believed to have the oldest ash.
The gravimetric water content of both ash types was determined by oven drying the ash at 105˚C for 24 h. After drying, weathered fly ash samples were manually ground and sieved to less than 2 mm. Fresh samples did not need to be ground and sieved. Pre-extraction pH and electrical conductivity (EC) of the ash samples were measured using a 1:10 solid/solution ratio of 10 g of fly ash and 100 mL of deionized (DI) water. Organic matter was determined by loss-on-ignition by oven drying the ash at 750˚C for 1 hr. Fresh and weathered fly ash acid- recoverable Se, As, Cr, Sb, Ba, Be, Cd, Pb, Hg, Ni, Ag, and V were determined following EPA Method 200.7/ 6010B at the AEP Analytical Chemistry Services Laboratory (Shreveport, LA) using a Varian VistaPro inductively coupled plasma-optical emission spectrometer (ICP-OES; Agilent Technologies, Santa Clara, CA) [45] . Chemical composition (e.g., silicon dioxide, aluminum oxide, iron oxide, calcium oxide, magnesium oxide, sulfur trioxide, sodium oxide, and potassium oxide) of the fly ash samples was determined by the Analytical Testing Service Laboratories (Joplin, MO) using American Society for Testing and Materials (ASTM) standard method C-311 [9] . Fresh and weathered fly ash sample analyses were conducted in triplicate.
The Applied Speciation and Consulting, LLC (Bothell, WA) laboratory also performed analyses on the fresh and weather ash used in the extraction procedure to partition the oxidation states of Se present [i.e., selenite 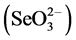 and selenate
and selenate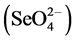 ]. Four replications of each ash type were analyzed, following a 2-hr alkaline extraction based on previous research by Jackson and Miller [46] , using ion chromatography, inductively coupled plasma, collision-reaction-cell, mass spectrometry (IC-ICP-CRC-MS) performed on an Agilent 7700 Series Inductively Coupled Plasma Mass Spectrometer (ICP-MS; Agilent Technologies, Santa Clara, CA).
]. Four replications of each ash type were analyzed, following a 2-hr alkaline extraction based on previous research by Jackson and Miller [46] , using ion chromatography, inductively coupled plasma, collision-reaction-cell, mass spectrometry (IC-ICP-CRC-MS) performed on an Agilent 7700 Series Inductively Coupled Plasma Mass Spectrometer (ICP-MS; Agilent Technologies, Santa Clara, CA).
2.3. Water Extractant Collection and Characterization
Rainwater was collected from an agriculturally managed pasture in Washington County, AR during a single storm event, using multiple plastic containers. Groundwater was collected from an uncontaminated, private well located adjacent to the ash landfill at a depth of approximately 61 m. Deionized water was collected from the outlet of the Flint Creek demineralizer. The Flint Creek demineralizer consists of a strong-acid cation and strong-base anion bed, followed by a mixed-bed polishing unit. Outflow from this design consistently produces ion concentrations of <1 µg∙L−1. Water extracts were stored at 4˚C until the Se, As, and Cr concentrations of the extractant solution could be determined in triplicate by the Environmental Testing Group (ETG) laboratory in Bentonville, AR, which is an Arkansas Department of Environmental Quality certified laboratory. The pH and EC were measured potentiometrically in-house at the Flint Creek laboratory. The water-soluble fractions of Se, As, and Cr were also calculated based on their respective initial measured concentrations in both the fresh and weathered ashes.
2.4. Ash Extraction
Water extractions were performed in 50-mL centrifuge tubes with a 10:1 extractant-to-fly ash dilution ratio (i.e., 30 mL extractant to 3 g fly ash) for all three water-extractant types at ambient room temperature, which was consistent with the procedures of previous studies [47] - [49] . Extractions were conducted for 2 and 6 hours using a 30-revolutions-per-minute (rpm), end-over-end shaker. Post-extraction pH and EC were measured potentiometrically before centrifuging the samples at 6000 rpm (11.5 G) for 10 minutes, followed by gravity filtering through a 0.45-µm Suppor-450 membrane filter (Pall Life Science, Port Washington, NY).
The filtrate was then preserved with Fisher® Optima Grade nitric acid to maintain a pH of less than 2. Samples were stored at 4˚C until shipment for chemical analysis could occur. A total of 60 (e.g., two fly ash types, three water extractants, two extraction times, and five replications) treatment samples and 12 treatment blanks (e.g., three water extractants, two extraction times, and two replications) were sent to the ETG laboratory for Se, As, and Cr analyses using an Agilent 7500 Series Inductively Coupled Plasma Mass Spectrometer (ICP-MS; Agilent Technologies, Santa Clara, CA). The United States EPA Method 200.8/6020 was used as the procedure guidelines for the proper calibration and analysis of the initial water extractants and water-extracted-ash solutions [50] [51] .
2.5. Statistical Analyses
A two-sample t-test was conducted to determine differences between initial fresh and weathered ash sample properties using Minitab (version 13.31, Minitab Inc., State College, PA). A one-factor analysis of variance (ANOVA) was conducted using SAS (version 9.3, SAS Institute, Inc., Cary, NC) to compare differences in initial water-extractant characteristics and to identify possible contamination of treatment blanks. A two-factor ANOVA was conducted to determine the effects of ash type, Se species, and their interaction on initial ash Se concentrations. A three-factor ANOVA was conducted to evaluate the effects of ash type, extraction time, water- extractant type, and their interaction on suspension pH and EC and Se, As, and Cr concentrations in the water- extracted ash samples. When appropriate, means were separated by least significant difference (LSD) at the 0.05 level. Linear correlations were also performed to evaluate the effects of water extractant, extractant property, and extraction time on the relationship between suspension pH and EC, and water-soluble Se, As, and Cr concentrations for the weathered ash using Microsoft Excel (version 2010, Microsoft Corporation, Redmond, WA).
3. Results and Discussion
3.1. Initial Ash Characteristics
Several initial chemical and physical characteristics differed between the two ash types (i.e., fresh and weathered). The fresh ash had lower water content (<0.01 g∙g−1) compared to the weathered ash (0.36 g∙g−1; p < 0.001; Table 2), which was expected due to the differences in degree of environmental exposure each ash had experienced. Fresh ash was collected from the outlet piping of the ESPs where the fresh ash had been exposed to temperatures of approximately 400˚C, while weathered ash was exposed to environmental weathering in the ash landfill. Organic matter differed (p < 0.001) between the fresh (0.39%) and weathered ash (6.4%; Table 2), which is consistent with the Class C fly ash organic matter concentration range of 0.3% to 17.5% reported by Lou et al. [5] . At Flint Creek, normal plant operation results in a fresh ash organic matter concentration that is consistently below 1.0%. Furthermore, exposure to environmental conditions (i.e., wind-blown organic matter as dust) at the landfill could contribute to the increased organic matter of the weathered ash. However, differences in initial ash organic matter concentration were not suspected to have affected the water extractabilities of trace metals because research by Lopez-Anton et al. [52] showed that organic matter in fly ash does not significantly influence As or Se adsorption/retention onto the fly ash.
As expected, pH differed (p < 0.001) 0.4 units between the fresh (pH = 11.6) and weathered ash (pH = 11.2; Table 2). Previous studies [53] [54] investigating eastern bituminous, western sub-bituminous, low-sulfur bituminous, sub-bituminous, lignite, and subbituminous PRB fly ash reported that the most alkaline (pH = 11.05 to 12.57) ash suspension was from sub-bituminous PRB ash. Environmental weathering and leaching of Ca, Fe, and S may have contributed to the lower pH of the weathered ash, which is supported by previous findings that fly ash with a Fe/Ca ratio > 3 produces acidic suspensions, while ratios < 3 generate alkaline suspensions [55] . The fresh and weathered ash had Fe/Ca ratios of 0.21 and 0.20, respectively. Furthermore, fly ashes with Ca/S ratios < 2.5 produce acidic suspensions, while ratios > 2.5 generate alkaline suspensions [16] . The fresh and weathered ash had Ca/S ratios of 19.3 and 17.2, respectively. Both the Fe/Ca and Ca/S ratio comparisons supported the observations that weathering and subsequent leaching likely resulted in a lower pH for the weathered ash compared to the fresh ash.
Table 2. Two-sample t-test summary of the initial ash characteristics used in the laboratory extractions. Mean [n = 3; ±standard error (SE)] values are reported.
*Significant at the 0.05 probability level. **Significant at the 0.01 probability level. ***Signifi- cant at the 0.001 probability level. aValues were below detection limits, and therefore no SE is reported.
Similarly, suspension EC differed (p < 0.001) between ash types, with EC being approximately two times greater for the fresh than weathered ash (Table 2), likely due to the leaching of major ash constituents such as Na, 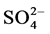 , and B(OH)4− from the weathered ash, as a result of environmental exposure. Due to the effects of time and exposure to water, major constituents can be released from the ash when exposed to water percolating downward through the ash. Comparing EC between studies can be difficult due to the wide array of solid/solu- tion ratios, agitation times, and initial elemental concentrations; therefore, the EC difference between fresh and weathered ash is likely more significant than the magnitude of the EC itself.
, and B(OH)4− from the weathered ash, as a result of environmental exposure. Due to the effects of time and exposure to water, major constituents can be released from the ash when exposed to water percolating downward through the ash. Comparing EC between studies can be difficult due to the wide array of solid/solu- tion ratios, agitation times, and initial elemental concentrations; therefore, the EC difference between fresh and weathered ash is likely more significant than the magnitude of the EC itself.
As expected, there were several differences in the oxide contents between ash types. Due to the suspected leaching and formation of secondary ash products, ferric oxide (p < 0.01), magnesium oxide (p < 0.001), and sodium oxide (p < 0.001) contents were lower for the weathered than fresh ash (Table 2). The sulfur trioxide content also differed (p < 0.05) between ash types; however, the weathered ash had a greater sulfur trioxide content (1.53%) than the fresh ash (1.39%; Table 2). Unexpectedly, the calcium oxide content did not differ between ash types and averaged 26.6% (Table 2). Silica was also greater (p < 0.01) in the fresh (34.9%) than in the weathered ash (32.0%; Table 2). Results from this study are consistent with the ASTM classification of Class C fly ash, which requires a minimum of 50% SiO2, Al2O3, and Fe2O3 and 20% CaO, compared to Class F fly ash, which has a minimum SiO2, Al2O3, and Fe2O3 content of 70% and normal CaO content of <10% [9] .
Total acid-extractable Se and As concentrations did not differ between ash types and averaged 4.0 mg Se kg−1 and 6.4 mg As kg−1 (Table 2). These concentrations were similar to sub-bituminous PRB ash concentrations reported by Lou et al. [5] and Seshadri et al. [56] , which ranged from 2.2 to 15 mg Se kg−1and from 1.8 to 27 mg As kg−1. In comparison, EPRI [16] reported mean bituminous ash Se and As concentrations of 12.3 and 219 mg∙kg−1, respectively. Coals with large sulfur concentrations (i.e., bituminous) are expected to contain greater As concentrations compared to low-sulfur coals (i.e., sub-bituminous PRB) because As is associated with Fe sulfides and other sulfide minerals in the coal [53] . In contrast, the acid-extractable Cr concentration was lower (p < 0.05) in the fresh (49.2 mg∙kg−1) than in the weathered ash (52.0 mg∙kg−1; Table 2), which was similar to that for sub-bituminous ash Cr reported by EPRI [16] , with a mean of 73 mg∙kg−1 and a range from 41 to 108 mg∙kg−1. Other acid-extractable metals that differed (p < 0.05) between ash types were Be, Cd, Pb, Hg, and V (Table 2). Unexpectedly, of these metals (i.e., Be, Cd, Pb, Hg, and V), only Be had a greater concentration in the fresh compared to the weathered ash.
Greater acid-extractable Cr concentration in the weathered than fresh ash may be due to the coal mining location and/or combustion conditions that produced the ash [5] . Even if the coal was from the same mine, differences may exist between the coal seams. Similarly, varying combustion conditions could affect trace metal release and re-precipitation onto the surface of the fly ash. Therefore, when trying to explain differences in trace metal concentrations between fresh and weathered ash, not only should leaching of trace elements be considered, but also factors such as coal source (i.e., differential coal seams) and combustion conditions. Furthermore, trying to compare ash trace metal concentrations among different studies can also be difficult because of the differences in power plant operating systems. For example, the coal source type (i.e., lignite, sub-bituminous, bituminous, or anthracite), furnace design (i.e., wall-, tangential-, or cyclone-fired furnace), combustion conditions (i.e., combustion temperature, flue-gas duct temperature, ash/element reaction time, and post-combustion oxygen concentration), and differences in equipment design/environmental upgrades (i.e., cold- or hot-side ESPs, wet- or dry-ash handling system, activated-carbon injection, wet- or dry-scrubber, and selective catalytic reduction equipment) can have a significant effect on initial ash characteristics.
Another factor that may contribute to initial ash variability, but one that is seldom mentioned, is the collection location and resulting differences in relative enrichment of Se onto the fly ash particles. As fly ash particles pass through multiple ESP fields, the ash particle size decreases resulting in increased fly ash surface area and increased Se adsorption [15] . The first two ESP fields of the Flint Creek ESP collect the majority of the larger diameter (i.e., 7.5 to 30 µm) fly ash particles, while fields three through eight capture the remaining ash particles (i.e., 3.5 to 7.5 µm). Therefore, as the fly ash passes through the eight fields of the Flint Creek ESP, greater Se adsorption, per volume of ash, should occur in the later fields. The first two fields will remove approximately 83% of the ash particles (Neundorfer Particulate Knowledge, personal communication, 2013); however, per volume, greater Se adsorption should be occurring in the later fields. Adsorption rates differing among ESP fields is important because if samples are collected from an ash hopper underneath one of the later fields, then Se may be greatly enriched compared to that collected from an earlier field. Andren and Klein [57] reported a fresh ash Se concentration of 28.0 µg∙kg−1 at the inlet of the precipitator and concentration of 88.3 µg Se kg−1 at the outlet. Therefore, reporting the exact fly ash collection location would provide useful insight in future studies. Collecting fly ash from an automated collection device installed on the outlet piping of the precipitator hoppers, or from a fly ash silo, may help reduce variability between initial trace metal concentrations in future studies.
As hypothesized, there were differences between fresh and weathered ash water-soluble selenite and selenate concentrations. Selenite was greater (p < 0.001) in the fresh (3.85 mg∙kg−1) than in the weathered ash (0.70 mg∙kg−1), while selenate was lower (p < 0.001) in the fresh (0.48 mg∙kg−1) than in weathered ash (0.67 mg∙kg−1; Table 2). Huggins et al. [58] reported similar results with selenite as the dominant form present in both fresh bituminous and sub-bituminous PRB ash. Comparing species across ash types, the concentration of selenite was greater (p < 0.001; 3.85 mg∙kg−1) than selenate (0.48 mg∙kg−1) in the fresh ash. In contrast, Se forms did not differ between weathered ash selenite and selenate (0.70 and 0.67 mg∙kg−1; respectively). Based on the assumption that all Se species were present as selenite and/or selenate, when compared to total-acid-extractable Se concentrations, the ash alkaline extraction recovered 103% of the fresh ash total Se, while only 36% of the weathered ash total Se was recovered. The difference between fresh and weathered ash Se recovery was likely due to the lack of selenite recovery in the weathered ash.
The differences between fresh and weathered ash selenite concentrations were likely due to the formation of ettringite [Ca6Al2(SO4)3(OH)12∙26H2O] and calcium selenite (CaSeO3) precipitate following hydration [58] -[60] . Wang et al. [47] similarly concluded that highly alkaline, washed ash acted as a sink for selenite through ettringite formation or precipitation with Ca; however, almost all of the selenate remained in the soluble phase. Baur and Johnson [60] reported that the precipitation of CaSeO3 limited the solubility of selenite through the formation of inner-sphere complexes, while selenate has a lower affinity for Ca-oxide surfaces and forms weakly bound, outer-sphere complexes. Furthermore, Baur and Johnson [60] and Solem-Tishmack et al. [61] reported that selenate was more readily leachable when bound to ettringite than selenite. Selenite sorption by ettringite was measured to be five times greater than sorption of selenate [60] .
Formation of secondary hydrated products (i.e., ettringite and CaSeO3) may take up to several months to allow equilibrium conditions to occur [62] . Therefore, hydration during the 2-hr alkaline extraction was not likely long enough to allow formation of the secondary hydrated products within the fresh ash that binds selenite, which allows the selenite to become solubilized during the alkaline extraction. However, the pre-hydrated weathered ash (water content = 0.36 g∙g−1; Table 2) would have likely stabilized the weathered ash ettringite and CaSeO3 and prevented solubilization of selenite [59] [63] .
3.2. Initial Water-Extractant Characteristics
As expected, there were several differences among initial water extractants (i.e., deionized water, rainwater, and groundwater) used for the laboratory extractions. The pH differed (p < 0.05) among all three water extractants with deionized water having the most alkaline pH (8.22), groundwater having a slightly alkaline, intermediate pH (7.64), and rainwater having the most acidic pH (5.24; Table 3). In addition to pH, though not explicitly measured in this study, the buffering capacity of these waters may be equally important. For example, the groundwater may contain a certain amount of Al or Fe, which would not only buffer the water to pH changes, but also may cause precipitation of amorphous Al or Fe hydroxide phases. Similarly, EC differed between all three water extractants with groundwater being approximately 336 times greater and rainwater 4.5 times greater than that of deionized water (Table 3). The significant differences with respect to EC suggest that particularly groundwater and rainwater have ion compositions that may be influencing the solubility of As, Cr and Se. However, Se, As, and Cr concentrations were all below instrument detection limits for all three water extractants (i.e., <0.005, <0.005, and <0.001 mg∙L−1, respectively; Table 3).
3.3. Treatment Effects on Extractable Fly-Ash Properties
All extractable fly-ash properties evaluated were affected by one or more treatment factors examined. Suspension pH and EC differed (p < 0.01) between ash types, among extraction times, and across extractant types (Ta- ble 4). In contrast to suspension pH and EC, water-soluble Se and As concentrations differed (p < 0.05) between ash types across extractant types, while water-soluble Cr concentration differed between ash types among extraction times (p < 0.01) and differed between ash types across extractant types (p = 0.02; Table 4).
As expected, fresh ash suspension pH and EC extracted with deionized water, rainwater, and groundwater for 2- and 6-hr were greater (p < 0.05) than for weathered ash extracted over the same treatments (Figure 1). Characteristics of the initial fresh and weathered ash, such as the Fe/Ca and Ca/S ratios, were likely responsible for the lower weathered ash suspension pH. Similarly, due to the suspected leaching of major elements from the weathered ash from environmental exposure, greater fresh-ash suspension EC was measured. Increasing the fresh and weathered ash extraction time from 2- to 6-hr across extractant types resulted in greater suspension pH and EC for all water extractants, except for the weathered ash EC extracted with groundwater, which did not differ between extraction times (Figure 1).
Fresh-ash suspension EC after 2 hours of extraction differed among all water extractants; EC averaged 2800 µS∙cm−1 from rainwater, 2710 µS∙cm−1 from deionized water, and 2500 µS∙cm−1 from groundwater (Figure 1). In contrast, the suspension EC for the weathered ash extracted for 2 hours did not differ (p > 0.05) among water extractants and averaged 1638 µS∙cm−1 (Figure 1). Both the fresh and weathered ash extracted with deionized water and rainwater for 6 hours resulted in similar suspension EC, but was 1.3 (fresh) and 1.1 (weathered) times greater than the suspension EC with groundwater after 6-hr extraction (Figure 1). The likely reason for differ-
Table 3. Analysis of variance summary of the initial water extractant characteristics used during the laboratory extractions. Mean [n = 3; ±standard error (SE)] values are reported. For the purpose of conducting statistical analyses, concentrations below instrument detection limits were reported as the detection limit value.
aMeans followed by different letters in a row are significantly different at the 0.05 level. bValues were below detection limits, and therefore no SE is reported.
Table 4. Analysis of variance summary of the effects of ash type, extraction time, extractant type, and their interactions on suspension pH and electrical conductivity (EC), and water-extractable selenium (Se), arsenic (As), and chromium (Cr) concentrations.
ences between 2- and 6-hr suspension EC was that extraction for 2 hours was not long enough to allow solubility equilibria. Furthermore, the combination of the low ionic strength of the deionized water and rainwater, the resulting gradient difference with the ash, the precipitation of certain elements originally present in the groundwater (e.g., Ca) with the extracted ions, and/or the common ion effect preventing dissolution with groundwater altogether could possibly explain the greater suspension EC when deionized water or rainwater were used compared to groundwater.
Evaluating the fresh ash extracted for 2 hours, the suspension pH was lower (p < 0.05) when rainwater (pH = 11.84) and groundwater (pH = 11.82), which did not differ, were used as the extractant compared to deionized water (pH = 11.89; Figure 1). Similar to suspension EC, suspension pH for fresh ash extracted for 6 hours was similar between extraction with deionized water and rainwater, but was 0.14 (deionized water) and 0.15 (rainwater) pH units greater than extraction with groundwater (Figure 1). When comparing among water-extractant types for the 2-hr extraction of weathered ash, suspension pH was also greater with deionized water (pH = 11.41) and rainwater (pH = 11.40) as the extractant type, which did not differ, compared to extraction with groundwater (pH = 11.31; Figure 1). Weathered ash extracted for 6 hours resulted in a slightly decreased suspension pH when deionized water (pH = 11.55), rainwater (pH = 11.51), and groundwater (pH = 11.41) were used (Figure 1). The resulting pH differences between extraction durations may be related to ash wetting period, where the ash may not have been completely wetted after 2 hours, but was after 6 hours. It appears that deionized water should be used for any combination of ash type and extraction time to obtain maximum suspension pH. Howev-

Figure 1. Suspension pH and electrical conductivity (EC) as affected by ash type (fresh and weathered), extraction time (2 and 6 hr), and water- extractant type [deionized water (DI), rainwater (RW), and ground-water (GW)]. Different letters atop bars within a property are significantly dif- ferent at the 0.05 level.
er, to ensure maximum fresh and weathered ash suspension pH and EC, it is recommended to perform a 6-hr extraction with deionized water even though several of the treatment factors did not differ between extraction with deionized water and rainwater. Using deionized water should ensure maximum suspension pH and EC and is more readily available than rainwater. Therefore, when focused exclusively on pH and EC, extraction with environmentally representative rainwater and groundwater offered no distinct benefits compared to deionized water as the extractant of choice.
Averaged over extraction times, weathered ash Se and As concentrations differed among water-extractant types, while fresh ash Se and As concentrations did not differ between water-extractant types (Figure 2). Fresh ash extractable Se and As concentrations were reported at or near the minimum detection limit (MDL) for all three water-extractant types, possibly explaining the lack of difference between the three water-extractant types. The extractable As concentration in the weathered ash was greater (p < 0.05) with rainwater (0.45 µg∙L−1) than with deionized water (0.36 µg∙L−1), while the groundwater-extractable As concentration was intermediate (0.41 µg∙L−1) and similar (p > 0.05) to that from both rainwater and deionized water (Figure 2). In contrast, the extractable Se concentration from the weathered ash was greater (p < 0.05) with groundwater (60.1 µg∙L−1) and rainwater (59.8 µg∙L−1), which did not differ, than extraction with deionized water (57.6 µg∙L−1; Figure 2). The Se and As results are consistent with previous research by Wang et al. [63] who used a 10:1 dilution ratio of deionized water and sub-bituminous ash and reported that As was not detected under most experimental factors and that Se was more readily extractable than As.
When comparing extractable As between ash types and across water-extractant types, the reported results could be misleading because all of the mean extractable As concentrations were <0.5 µg∙L−1 (Figure 2). Since these concentrations are at or near the MDL of 0.2 µg∙L−1, it may be difficult to differentiate and compare reported As results with any degree of confidence. Wang et al. [63] reported that As did not leach from sub-bitu- minous ash until a 2:1 deionized water dilution ratio was used for a total of 30 d using a 180-rpm shaker. There-

Figure 2. Water-soluble selenium (Se), arsenic, (As), and chromium (Cr) concentration differences between ash types (fresh and weathered) across water-extractant types [deionized water (DI), rainwater (RW), and groundwater (GW)], averaged over extraction times. Different letters atop bars within a property are significantly different at the 0.05 level.
fore, a longer extraction time and lower dilution ratio may be required to obtain more relevant data pertaining to water-soluble As concentrations.
Averaging over water-extractant types, weathered ash water-soluble Cr concentration increased as the extraction increased from 2 (166.9 µg∙L−1) to 6 hours (174.7 µg∙L−1; Figure 3). For comparison, EPRI [64] reported that the water-soluble Cr fraction over a one-week laboratory extraction was 42.3% (0.25 hr), 42.3% (24 hr), 11.5% (48 hr), and 3.9% (168 hr) using a 10:1 dilution ratio. Therefore, it is recommended that, if Cr is the trace metal of interest, extraction occur for at least 24 hours to extract the majority of the water-soluble Cr from weathered ash. Water-soluble Cr concentration in the fresh ash was unaffected by extraction time and averaged 1.2 µg Cr L−1 (Figure 3). Similar to Se and As, fresh ash water-soluble Cr concentration, averaged over extraction times, did not differ between water-extractant types as would be expected since measured concentrations were at or near the MDL (Figure 2). However, for the weathered ash, extraction with groundwater resulted in a greater (p < 0.05) extractable Cr concentration (176.3 µg Cr L−1) compared to that from rainwater (170.1 µg Cr L−1) and deionized water (166.0 µg Cr L−1), which did not differ (Figure 2).
Possible reasons for why weathered ash Se and Cr extraction was greatest with groundwater include differences in suspension pH and EC between the water extractants (Figure 1). Selenium and Cr can be incorporated into ettringite by substitution with the sulfate ion at pH > 11.5, but become unstable at a pH < 10.7 [24] [62] . Weathered ash extraction with groundwater resulted in the lowest suspension pH when compared to rainwater and deionized water (Figure 1). Therefore, a greater amount of Se or Cr could have been incorporated into the ettringite, due to the greater weathered ash suspension pH from extraction with deionized water and rainwater. The ettringite thus acts as a sink for Se or Cr and prevents solubilization [47] [59] .
The 6-hr extraction with deionized water and rainwater resulted in greater weathered ash suspension EC compared to that from groundwater (Figure 1). However, there was no difference in suspension EC between the

Figure 3. Water-soluble chromium (Cr) concentration differences between ash types (fresh and weathered) among extraction times (2 and 6 hr), averaged over water-extractant types. Different letters atop bars are significantly different at the 0.05 level.
water extractants for the 2-hr extraction. Increased suspension EC resulted from the greater concentration of ions in solution. However, more evidence is needed to determine why greater weathered ash Se and Cr extraction occurred with environmentally representative groundwater. Weathered ash suspension pH and EC did not differ between rainwater and deionized water, except for the 6-hr extraction suspension pH (Figure 1).
From these results, it is clear that a significant difference existed between fresh and weathered ash Se and Cr water-soluble concentrations, with possible differences between fresh and weathered ash water-soluble As concentrations. Information regarding oxidation state is essential in understanding and predicting Se, As, and Cr behavior and may help explain extraction differences between ash types [53] . For example, selenite, arsenate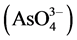 , and trivalent Cr are known to be less water-soluble than selenate, arsenite
, and trivalent Cr are known to be less water-soluble than selenate, arsenite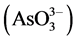 , and hexavalent Cr [27] .
, and hexavalent Cr [27] .
When examining redox-active species, the initial form within the coal is not important because virtually all of the Se, As, and the majority of the Cr is volatized into the gas phase during combustion; therefore, the redox-ac- tive form in the initial coal has no effect on the post-combustion form [58] . As fly ash weathers, trace metal redox-active forms may also change, resulting in a change in leaching behavior. For example, selenate is very soluble, while selenite is only sparingly soluble and is strongly adsorbed onto hydrous oxide surfaces. EPRI [27] reported that Se in weathered sub-bituminous ash landfill leachate was almost entirely in the selenate form. However, Wang et al. [63] reported that fresh sub-bituminous PRB ash exhibited low leaching potential, which is consistent with Se in the selenite form; therefore, the field-derived data may indicate conversion from selenite to selenate from environmental exposure in the landfill. Though the reaction kinetics of the oxidation of selenite to selenate are not well-understood, this reaction appears to be a slow process [65] . Oxidation state may also explain the differences noted between fresh and weathered ash water-soluble Se concentrations. Assuming that all Se was present as selenite or selenate, fresh ash Se was predominately in the less-water-soluble selenite form (88.9%), compared to selenate (11.1%), likely explaining why fresh ash water-extractable Se concentrations were at or below MDL for all three water-extractants (Figure 2). Similarly, Narukawa et al. [25] reported fresh ash water-soluble Se concentrations were present predominately as selenite, but the coal source was not reported.
Mattigod and Quinn [66] reported that selenite from sub-bituminous ash was completely oxidized to selenate after one week using a 4:1 dilution ratio. In contrast, Wang et al. [63] reported that an extraction time of 30 d had no effect on Se conversion from selenite to selenate under natural conditions (i.e., exposed to air), regardless of dilution ratio. Therefore, in some instances, 30 d may not be enough exposure to stimulate a change in Se oxidation state. Environmental weathering in the landfill may have affected Se oxidation state, promoting the oxidation of selenite to selenate. The highly water-soluble selenate concentration was greater (p < 0.001) in the weathered (0.67 mg∙kg−1) than the fresh ash (0.48 mg∙kg−1), while the selenite concentration was less (p < 0.001) in the weathered (0.70 mg∙kg−1) than in the fresh ash (3.85 mg∙kg−1). Therefore, oxidation state likely explains the greater water-extractable weathered ash Se concentrations across all three water-extractants (Figure 2).
Wang et al. [63] reported no significant change in speciation of As during a 30-d extraction. Wang et al. [63] also reported non-detectable As concentrations for the fresh sub-bituminous PRB ash except with a 2:1 dilution ratio and 30-d extraction, which resulted in water-soluble As concentrations of 20 µg∙L−1, with As primarily in the arsenate form. Narukawa et al. [25] reported fresh ash water-soluble As concentrations were dominated by the less mobile and less toxic arsenate form, but did not report the coal source. Huggins et al. [58] reported similar results with arsenate as the dominant form present in both fresh bituminous and sub-bituminous PRB ash, thereby likely explaining the fresh ash water-extractable As concentrations being at or the near the MDL across all three extractants.
The low water-extractable weathered ash As concentrations may be explained by the large calcium content of the ash, which may have caused As precipitation with the Ca, or substitution with ettringite. Selenium, As, and Cr can be incorporated (i.e., substituted) into ettringite at pHs > 11.5, but become unstable at a pH < 10.7 [24] , which is consistent with measured suspension pH values from this study (Figure 1). However, ettringite formation has been reported by Hassett et al. [67] as a long-term process (i.e., >18 hr) between water and ash; therefore, fresh ash extraction should not have been affected by ettringite formation because the fresh ash was not exposed to water prior to extraction in this study.
Differences in oxidation states may also explain increased water-extractability of Cr from the weathered ash. In fresh ash samples, Stam et al. [68] reported that the less mobile trivalent Cr was the dominate form present. However, the alkaline pH of sub-bituminous PRB ash suspensions promotes conversion of trivalent Cr to the more water-soluble hexavalent Cr [69] [70] . Similarly, in a study of 29 landfill leachates, EPRI [27] reported hexavalent Cr as the dominant form present when pH was greater than 6, with concentrations of 0.5 to 5100 µg Cr L−1.
Although the redox status of Cr and As were not directly determined, differences between fresh and weathered ash water-soluble Cr concentrations indicate that the less water-soluble trivalent Cr was likely dominant in the fresh ash and the more water-soluble hexavalent Cr was likely dominant in the weathered ash. Weathered ash water-soluble Se and Cr concentrations indicate that there is still a significant amount of Se and Cr readily available to be released into the environment if allowed to come in contact with rainwater or groundwater. Fresh and weathered ash water-soluble As concentrations at or near the MDL were likely due to As existing and remaining in the arsenate form. Previous research indicated that the less water-soluble arsenate is the dominate form present in fresh sub-bituminous PRB ash and it is unlikely that the arsenate is being reduced to the more water-soluble arsenite in the landfill [58] . Therefore, results from this study indicate that redox status is likely responsible for the differences between extraction behavior of fresh and weathered ash Se, As, and Cr concentrations.
3.4. Water-Soluble Se, As, and Cr Fractions
To better characterize fly ash trace metals, Narukawa et al. [25] and Yuan [26] performed a chemical fractionation that distributed trace metals into five fractions: water-soluble, acid-soluble (i.e., bound to carbonates; extracted with acetic acid), reducible (i.e., bound to Fe-Mn oxides), oxidizable (i.e., bound to organic matter), and residual (i.e., bound to silica; extracted with HNO3 and HCl). Of these, it was concluded that the water-soluble fraction is the most readily available fraction that can be easily released into the environment through contact with rainwater and groundwater [25] [26] . Averaged over extraction time and extractant type, the total water-soluble fractions of Se, As, and Cr were determined for fresh and weathered ash. The fresh ash water-soluble fractions for Se (0.05%), As (<0.01%), and Cr (<0.01%) were very low since concentrations were at, or near, MDL values. The weathered water-soluble fractions for Se, As, and Cr were 1.6, 0.01, and 0.33%, respectively, which were lower than reported fresh-ash results by Narukawa et al. [25] and Yuan [26] . However, ash samples from Narukawa et al. [25] and Yuan [26] were collected at eight different locations throughout the world and exhibited different initial ash characteristics than those in this study.
3.5. Correlations among Ash Properties
Linear correlations were used to evaluate the effects of water extractant, extractant property, and extraction time on the relationship between suspension pH and EC and water-soluble Se, As, and Cr concentrations. Only wea-
Table 5. Summary of the linear correlations (r) between suspension pH and EC, and water-soluble selenium (Se), arsenic (As), and chromium (Cr) concentrations from the 2- and 6-hr extraction times of the weathered ash using distilled water (DI), rainwater (RW), and groundwater (GW).
*Significant at the 0.05 probability level. **Significant at the 0.01 probability level. ***Signifi- cant at the 0.001 probability level. aElectrical conductivity.
thered ash data were analyzed since the fresh ash water-soluble Se, As, and Cr concentrations were all reported at, or near, the MDL. Water-soluble Se concentration was positively correlated (r = 0.95; p < 0.01) with suspension EC for weathered ash extracted for 6 hours with rainwater (Table 5). Similarly, weathered ash water-so- luble Se concentrations was positively correlated (r = 0.96; p < 0.01) with suspension pH for the 2-hr extraction with deionized water (Table 5).
In contrast to Se, weathered ash As and Cr concentrations were unrelated to suspension pH. Weathered ash water-soluble As concentration was inversely related (r = −1.00; p < 0.001) to suspension EC for the 2-hr extraction with rainwater (Table 5); however, four of the five replicate concentrations were 0.5 µg∙L−1, with the other being 0.4 µg∙L−1, possibly skewing the data. Therefore, in contrast to the statistical data, no linear relationship between water-soluble As and suspension EC was suspected. Extraction for 2 hours with deionized water resulted in an inverse relationship (r = −0.93; p < 0.05) between weathered ash water-soluble Cr concentration and suspension EC (Table 5).
Although some of the data appeared to support a linear relationship between weathered ash properties for various treatments (i.e., water extractant and extraction time), several underlying factors may be skewing these results since there appears to be little to no consistency within or across treatment combinations. For example, previous research EPRI [27] and Wang et al. [47] reported a strong linear relationship between water-soluble Se and pH; however, these data were collected over a wide pH range (i.e., pH 2 to 12), while site-specific, weathered ash suspension pH from this study only varied by 0.26 units.
4. Summary and Conclusions
Fresh and weathered sub-bituminous PRB fly ash had significantly different water-extractable Se, As, and Cr characteristics, with fresh ash water-soluble concentrations at or near the MDLs across all treatments. Differences between fresh and weathered ash were likely due to differences in the redox status of elements in the two ashes. Selenite was shown to be the dominate form present in the fresh ash, likely preventing the release of Se during the water-extraction study due to its low solubility. Once land-filled, environmental weathering likely promotes the oxidation of selenite to the highly mobile, water-soluble selenate.
Weathered ash water-soluble Se concentrations were greatest when extraction occurred with environmentally representative rainwater and groundwater. Similarly, weathered ash water-extractable Cr concentration was greatest with groundwater. Selenium and As extraction was unaffected by extraction time. In contrast, the weathered ash water-soluble Cr concentration increased as the extraction time increased from 2 to 6 hr. However, this research was not able to demonstrate if 6 hours were enough time to ensure maximum Cr extraction; therefore, a 24-hour extraction is recommended for Cr. Water-soluble As concentrations were too near the MDL to make an informed decision for both fresh and weathered ash. Therefore, a longer extraction time and lower dilution ratio may be required to obtain measurable water-extractable As concentrations from a Class C fly ash. To obtain representative fresh and weathered ash suspension pH and EC, a 6-hr or longer extraction with deionized water for both fresh and weathered ash is recommended. Extraction with environmentally representative rainwater and groundwater offered no distinct benefits compared to deionized water as the extractant of choice for suspension pH and EC. Results from this study were derived from the extraction of a Class C fly ash produced from the combustion of a sub-bituminous PRB coal and may not be comparable to extraction from Class F fly ash.
References
- United States Energy Information Administration (USEIA) (2011) Electric Power Annual. http://www.eia.doe.gov/
- Tripodi, R.A. and Cheremisinoff, P.N. (1980) Coal Ash Disposal: Solid Waste Impacts. Technomic Publishing Com- pany, Westport.
- Fizette, H.H. (2005) Development of Concrete Composites by Synergistically Using Illinois PCC Bottom Ash and Class C Fly Ash. PhD. Diss. Southern Illinois University, Carbondale.
- Sayiri, S.R.K. (2005) Utilization of Wyoming Bottom Ash in Asphalt Mixes. PhD Dissertation, University of Wyom- ing, Laramie.
- Lou, Y., Giammar, D.E., Huhmann, B.L. and Catalano, J.G. (2011) Speciation of Selenium, Arsenic, and Zinc in Class C Fly Ash. Energy & Fuels, 25, 2980-2987.
- Electric Power Research Institute (EPRI) (1984) Testing and Correlation of Fly Ash Properties with Respect to Pozzo- lanic Behavior. CS-3314. http://www.epri.com
- Wesche, K. (1991) Fly Ash in Concrete: Properties and Performance. E & FN Spoon, Great Britain.
- Yeheyis, M.B. (2008) Evaluation of Coal Fly Ash for Use in Mine Waste Management. PhD. Dissertation, University of Western Ontario, Canada.
- American Society for Testing and Materials (ASTM) (2006) Standard Test Methods for Sampling and Testing Fly Ash or Natural Pozzolans for Use in Portland-cement Concrete: ASTM C311. http://www.astm.org.standards/C311.htm
- American Coal Ash Association (ACAA) (2011) Corrected 2009 Coal Combustion Product (CCP) Production and Use Survey. http://acaa.affiniscape.com/associations/8003/files/2009_CCP_Production_Use_Survey_Corrected_020811.pdf
- Helmuth, R.A. (1987) Fly Ash in Cement and Concrete. Portland Cement Co., Skokie.
- Kiwerska, K.N. (2002) Crushed Aggregates from Class C Fly Ash. Ph.D. Dissertation, University of Missouri, Kansas City.
- Capp, J.P. and Spencer, J.D. (1970) Fly Ash Utilization: A Summary of Applications and Technology. United States Bureau of Mines, Information Circular, 8483, 1-72.
- Martens, D.C., Schnappinger, M.G., Doran, J.W. and Mulford, F.R. (1970) Ash Utilization: Fly Ash as a Fertilizer. United States Bureau of Mines, Information Circular, 8488, 310-330.
- Huang, Y., Jin, B., Zhong, Z., Xiao, R., Tang, Z. and Ren, H. (2004) Trace Elements (Mn, Cr, Pb, Se, Zn, Cd and Hg) in Emissions from a Pulverized Coal Boiler. Fuel Processing Technology, 86, 23-32. http://dx.doi.org/10.1016/j.fuproc.2003.10.022
- Electric Power Research Institute, EPRI (1987) Chemical Characterization of Fossil Fuel Wastes. EA-5321. http://www.epri.com
- Lemly, A.D. (1999) Selenium Impacts on Fish: An Insidious Time Bomb. Human and Ecological Risk Assessment, 5, 1139-1151. http://dx.doi.org/10.1080/10807039.1999.10518883
- Spallholz, J.E. and Hoffman, D.J. (2002) Selenium Toxicity: Cause and Effects in Aquatic Birds. Aquatic Toxicology, 57, 27-37. http://dx.doi.org/10.1016/S0166-445X(01)00268-5
- Chapman, P.M., Adams, W.J., Brooks, M.L., Delos, C.G., Luoma, S.N., Maher, W.A., Ohlendorf, H.M., Presser, T.S. and Shaw, D.P. (2009) Ecological Assessment of Selenium in the Aquatic Environment: Summary of a SETAC Pells- ton Workshop. http://wwwrcamnl.wr.usgs.gov/Selenium/Library_articles/pellston_SELSummary.pdf
- World Health Organization, WHO (1996) Trace Elements in Human Nutrition and Health. http://www.who.int/nutrition/publications/micronutrients/9241561734/en/index.html
- Environmental Protection Agency, EPA (2011) Ground Water and Drinking Water. http://water.epa.gov/drink/index.cfm
- Electric Power Research Institute, EPRI (2001) Evaluation of a Remedial Action at an Unlined Coal Ash Landfill: CS Site. TR-1005214. http://www.epri.com
- Electric Power Research Institute, EPRI (2002) Evaluation of a Remedial Action at an Unlined Coal Ash Landfill: PI Site. TR-1005262. http://www.epri.com
- Electric Power Research Institute, EPRI (2006) Weathering Processes and Secondary Minerals Formed in Coal Ash. IR-1012582. http://www.epri.com
- Narukawa, T., Takatsu, A., Chiba, K., Riley, K.W. and French, D.H. (2005) Investigation on Chemical Species of Ar- senic, Selenium and Antimony in Fly Ash from Coal Fuel Thermal Power Stations. Journal of Environmental Moni- toring, 7, 1342-1348. http://dx.doi.org/10.1039/b509817c
- Yuan, C.G. (2009) Leaching Characteristics of Metals in Fly Ash from Coal-fired Power Plant by Sequential Extrac- tion Procedure. Microchimica Acta, 165, 91-96. http://dx.doi.org/10.1007/s00604-008-0103-5
- Electric Power Research Institute, EPRI (2006) Characterization of Field Leachates at Coal Combustion Product Management Sites: Arsenic, Selenium, Chromium and Mercury Speciation. TR-1012578. http://www.epri.com
- Environmental Protection Agency, EPA (2004) Draft Aquatic Life Water Quality Criteria for Selenium. http://water.epa.gov/scitech/swguidance/standards/criteria/aqlife/selenium/upload/complete-2.pdf
- Elrashidi, M.A., Adriano, D.C., Workman, S.M. and Lindsay, W.L. (1987) Chemical Equilibria of Selenium in Soils: A Theoretical Development. Soil Science, 144, 141-152. http://dx.doi.org/10.1097/00010694-198708000-00008
- Goh, K.H. and Lim, T.T. (2004) Geochemistry of Inorganic Arsenic and Selenium in a Tropical Soil: Effect of Reaction Time, pH and Competitive Anions on Arsenic and Selenium Adsorption. Chemosphere, 55, 849-859. http://dx.doi.org/10.1016/j.chemosphere.2003.11.041
- Otero-Rey, J.R., Mato-Fernandez, M.J., Moreda-Pineiro, J., Alonso-Rodriguez, E., Muniategui-Lorenzo, S., Lopez-Mahia, P. and Prada-Rodriguez, D. (2005) Influence of Several Experimental Parameters on As and Se Leaching from Coal Fly Ash Samples. Analytica Chimica Acta, 531, 299-305. http://dx.doi.org/10.1016/j.aca.2004.10.029
- Ylaranta, T. (1982) Volatilization and Leaching of Selenium Added to Soils. Annales Agriculturae Fenniae, 21, 103-114.
- Ahlrichs, J.S. and Hossner, L.R. (1987) Selenate and Selenite Mobility in Overburden by Saturated Flow. Journal of Environmental Quality, 16, 95-98. http://dx.doi.org/10.2134/jeq1987.00472425001600020001x
- Barceloux, D. (1999) Selenium. Journal of Toxicology-Clinical Toxicology, 37, 145-172. http://dx.doi.org/10.1081/CLT-100102417
- Goldberg, S., Hyun, S. and Lee, L.S. (2008) Chemical Modeling of Arsenic (III, V) and Selenium (IV, VI) Adsorption by Soils Surrounding Ash Disposal Facilities. Vadose Zone Journal, 7, 1231-1238. http://dx.doi.org/10.2136/vzj2008.0013
- Neal, R.H. and Sposito, G. (1989) Selenate Adsorption on Alluvial Soils. Soil Science Society of America Journal, 53, 70-74. http://dx.doi.org/10.2136/sssaj1989.03615995005300010013x
- Peak, D. and Sparks, D.L. (2002) Mechanisms of Selenate Adsorption on Iron Oxides and Hydroxides. Environmental Science & Technology, 36, 1460-1466. http://dx.doi.org/10.1021/es0156643
- United States Energy Information Administration, USEIA (2010) Monthly Electric Usage/Home. http://www.eia.doe.gov/
- Wolniak, D.E., Haley, D.A., Millard, W., Tubbs, D.E., Stawasz, S.M. and Gutekunst, R.B. (1978) Engineering Data: Flint Creek Power Plant. Sargent & Lundy Engineers, Chicago.
- Sargent and Lundy Engineers (1974) Flint Creek Power Plant Environmental Report. Chicago.
- Kitto, J.B. and Stultz, S.C. (2005) Steam: Its Generation and Use: 41st Edition. Babcock & Wilcox Company, Barberton.
- Electric Power Research Institute, EPRI (1982) Electrostatic Precipitator Reference Manual. CS-2809. http://www.epri.com
- Terracon Consulting (Terracon) (2006) NOD Response to Major Permit Modification Revisions. http://www.adeq.state.ar.us/ftproot/Pub/WebDatabases/SolidWaste/PermittedFacilities/GenDocs/38963
- Arkansas Department of Environmental Quality, ADEQ (2010) Permit for the Construction and Operation of a Class 3N Solid Waste Disposal Facility: Permit 0273-S3N-R1. http://www.adeq.state.ar.us/ftproot/Pub//Solidwaste/PermittedFacilities/GenDocs/57847
- Environmental Protection Agency, EPA (2007) Inductively Coupled Plasma Atomic Emission Spectrometry: Method 6010C. http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/6010c.pdf
- Jackson, B.P. and Miller, W.P. (2000) Effectiveness of Phosphate and Hydroxide for Desorption of Arsenic and Selenium Species from Iron Oxides. Soil Science Society of America Journal, 64, 1616-1622. http://dx.doi.org/10.2136/sssaj2000.6451616x
- Wang, J., Wang, T. and Burken, J.G. (2007) The Leaching Characteristics of Selenium from Coal Fly Ashes. Journal of Environmental Quality, 36, 1784-1792. http://dx.doi.org/10.2134/jeq2007.0143
- McDonald, M., Brye, K.R., Miller, D.M. and Gbur, E.E. (2009) Broiler-Litter Application and Soil-Depth Effects on Arsenic Sorption. Soil Science, 174, 661-675. http://dx.doi.org/10.1097/SS.0b013e3181c5e4fb
- Daigh, A.L., Brye, K.R., Sharpley, A.N., Miller, D.M. and Gbur, E.E. (2010) Broiler Litter Composition as Affected by Water Extractant, Dilution Ratio and Extraction Time. Communications in Soil Science and Plant Analysis, 41, 2340-2357. http://dx.doi.org/10.1080/00103624.2010.508098
- Environmental Protection Agency, EPA (1994) Determination of Trace Elements in Waters and Wastes by Inductively Coupled Plasma-Mass Spectrometry. http://www.epa.gov/region9/qa/pdfs/200_8dqiI1.pdf
- Environmental Protection Agency, EPA (2007) Inductively Coupled Plasma Mass Spectrometry: Method 6020A. http://www.epa.gov/osw/hazard/testmethods/sw846/pdfs/6020a.pdf
- Lopez-Anton, M.A., Diaz-Somoano, M., Spears, D.A. and Martinez-Tarazona, M.R. (2006) Arsenic and Selenium Capture by Fly Ashes at Low Temperature. Environmental Science & Technology, 40, 3947-3951. http://dx.doi.org/10.1021/es0522027
- Electric Power Research Institute, EPRI (1998) Leaching of Inorganic Constituents from Coal Combustion By-Products under Field and Laboratory Conditions. TR-111773-V1. http://www.epri.com
- Electric Power Research Institute, EPRI (2012) Adsorption of Trace Elements on Fresh and Weathered Coal Fly Ash. TR-1023740. http://www.epri.com
- Mattigod, S.V., Rai, D., Eary, L.E. and Ainsworth, C.C. (1990) Geochemical Factors Controlling the Mobilization of Inorganic Constituents from Fossil Fuel Combustion Residues: I. Review of the Major Elements. Journal of Environmental Quality, 19, 188-201. http://dx.doi.org/10.2134/jeq1990.00472425001900020004x
- Seshadri, P., Sisk, D., Bowman, F., Benson, S. and Seames, W. (2011) Leachability of Arsenic and Selenium in Submicron Coal Fly Ash. World of Coal Ash Conference. http://www.flyash.info/
- Andren, A.W. and Klein, D.H. (1975) Selenium in Coal-fired Steam Plant Emissions. Environmental Science & Technology, 9, 856-858. http://dx.doi.org/10.1021/es60107a002
- Huggins, F., Senior, C.L., Chu, P., Ladwig, K. and Huffman, G.P. (2007) Selenium and Arsenic Speciation on Fly Ash from Full-Scale Coal-Burning Utility Plants. Environmental Science & Technology, 41, 3284-3289. http://dx.doi.org/10.1021/es062069y
- Lecuyer, I., Bicocchi, S., Ausset, P. and Lefevre, R. (1996) Physico-Chemical Characterization and Leaching of Desulphurization Coal Fly Ash. Waste Management Research, 14, 15-28. http://dx.doi.org/10.1177/0734242X9601400103
- Baur, I. and Johnson, C.A. (2003) Sorption of Selenite and Selenate to Cement Materials. Environmental Science & Technology, 37, 3442-3447. http://dx.doi.org/10.1021/es020148d
- Solem-Tishmack, J.K., McCarthy, G.J., Docktor, B., Eylands, K.E., Thompson, J.S. and Hassett, D.J. (1995) High- Calcium Coal Combustion Byproducts: Engineering Properties, Ettringite Formation and Potential Application in Solidification and Stabilization of Selenium and Boron. Cement and Concrete Research, 25, 658-670. http://dx.doi.org/10.1016/0008-8846(95)00054-G
- Hassett, D.J. and Pflughoeft-Hassett, D.F. (2002) Evaluating Coal Combustion By-Products (CCBs) for Environmental Performance. Proceedings of Coal Combustion By-Products and Western Coal Mines: A Technical Interactive Forum, Golden, 16-18 April 2002, 79-82. http://www.osmre.gov/programs/TDT/ccr/docs/CCB_and_Western_Coal_Mines.pdf
- Wang, T., Wang, J., Tang, Y., Shi, H. and Ladwig, K. (2009) Leaching Characteristics of Arsenic and Selenium from Coal Fly Ash: Role of Calcium. Energy & Fuels, 23, 2959-2966. http://dx.doi.org/10.1021/ef900044w
- Electric Power Research Institute, EPRI (1994) A Field and Laboratory Study of Solute Release from Sluiced Fly Ash. TR-104585. http://www.epri.com
- Seby, F., Potin-Gautier, M., Giffaut, E. and Donard, O.F.X. (1998) Assessing the Speciation and the Biogeochemical Processes Affecting the Mobility of Selenium from a Geological Repository of Radioactive Wastes to the Biosphere. Analusis, 26, 193-198. http://dx.doi.org/10.1051/analusis:1998134
- Mattigod, S.V. and Quinn, T.R. (2003) Selenium Content and Oxidation States in Fly Ashes from Western US Coals. In: Sajwan, K.S., Alva, A.K. and Keefer, R.F., Eds., Chemistry of Trace Elements in Fly Ash, Springer, New York.
- Hassett, D.J., Pflughoeft-Hassett, D.F. and Heebink, L.V. (2005) Leaching of CCBs: Observations from over 25 Years of Research. Fuel, 84, 1378-1383. http://dx.doi.org/10.1016/j.fuel.2004.10.016
- Stam, A.F., Meij, R., Winkel, H.T., Eijk, R.J., Huggins, F.E. and Brem, G. (2011) Chromium Speciation in Coal and Biomass Co-Combustion Products. Environmental Science & Technology, 45, 2450-2456. http://dx.doi.org/10.1021/es103361g
- Electric Power Research Institute, EPRI (1986) Geochemical Behavior of Chromium Species. EA-4544. http://www.epri.com
- Electric Power Research Institute, EPRI (2011) Chromium in Coal Combustion Products. TB-1022143. http://www.epri.com
- Emke, W.B. (2011) Rochelle/North Antelope: 2011 through 2015-Typical Analysis. http://coal.infomine.com/