International Journal of Clinical Medicine
Vol.07 No.05(2016), Article ID:66856,5 pages
10.4236/ijcm.2016.75036
A New Case of Turner Syndrome with Early Pubertal Development
Seok Ho Yoon, Dong Jun Lee, Son Moon Shin, So Young Yoon, Sung Won Park
Department of Pediatrics, Cheil General Hospital and Women’s Healthcare Center, College of Medicine, Dankook University, Seoul, Korea

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 30 March 2016; accepted 24 May 2016; published 27 May 2016
ABSTRACT
Turner syndrome (TS) is a relatively common chromosomal disorder caused by complete or partial X monosomy. The most frequently observed karyotype, 45, X, arises more often by loss of the paternal X or Y chromosome in meiosis or in early embryogenesis than by loss of the maternal X chromosome. The main clinical features of TS are short stature, gonadal dysgenesis, and not to undergo pubertal development (e.g. primary amenorrhea). However, a few rare cases of TS have shown precocious puberty. Our case of a 9-year-old girl did not have any TS-specific clinical hallmarks, with the exception of short stature. She visited our clinic because of her pubertal development and short stature. In this report, we highlight the variability that can occur in patients with TS and emphasizes the need to carefully assess unusual growth patterns in any child, regardless of other underlying conditions.
Keywords:
Turner Syndrome, Early Pubertal Development

1. Introduction
Turner syndrome (TS) is a relatively common chromosomal disorder caused by complete or partial X monosomy, with a prevalence of approximately 1/2000 in female live births [1] . Conventional karyotype analysis has revealed various genetic backgrounds of TS. The most frequently observed karyotypes are 45, X karyotypes with an isochromosome of X [i(Xq) or i(Xp)], the mosaic 45, X/46XX karyotype, and karyotypes containing an entire Y chromosome or parts thereof [2] . The mosaic TS karyotype occurs in approximately 30% of all patients with TS [3] . The main clinical features of TS are short stature, gonadal dysgenesis, and congenital malformations. Moreover, most individuals with TS do not undergo pubertal development and exhibit primary amenorrhea due to accelerated loss of oocytes in the 45, X ovary, leaving only a few follicles in the fibrous strike at birth. Approximately one-third of all girls with TS undergo spontaneous puberty, but only half of them complete puberty with menarche. Spontaneous pregnancies are rare (2% - 5%) [4] . We report a 9-year-old patient with early pubertal development who was later diagnosed with TS and karyotyped as 46, X, der (X) t (X; X) (p11.21; q11.2).
2. Case Report
A female child was born at a gestational age of 35 + 2 weeks with a birth weight of 1930 g (10th percentile on a Korean standard growth chart) and a length of 41.5 cm (10th percentile on a Korean standard growth chart). She had been admitted as a newborn in the nursery room with transient tachypnea. She had normal brain ultrasonography that was further evaluated due to her intrauterine growth retardation. During the newborn period, no signs of TS were manifested and no family history of genetic or congenital disorders was present. In 2012 when the patient was 7 years old, she visited another hospital because of high fever and was diagnosed with Kawasaki disease and Brown syndrome. She underwent annual echocardiograms, all of which were normal. When she was 9 years old, the patient was referred to our pediatric endocrinology outpatient clinic with complaints of short stature and breast budding. Her height on referral was 122.4 cm, which placed her in the 10th percentile on a Korean standard growth chart and at the 90th percentile on a Korean TS growth chart; her weight was 30.5 kg (70th percentile). The mid-parental height was 164.5 cm (75th percentile on a Korean standard growth chart). A physical examination revealed Tanner stage III breast development and Tanner stage I pubic hair development. Her bone age was 11 years. These clinical, radiologic, and laboratory findings were suspicious of idiopathic central precocious puberty; therefore, a GnRH stimulation test was performed. Due to her short stature, chromosomal screening was also performed.
Conventional chromosome analysis from blood lymphocytes revealed a karyotype of 46, X, der (X) t (X; X) (p11.21; q11.2) (Figure 1). A further screening test for TS was carried out. Kidney ultrasound results were normal and a pelvic ultrasound showed a prepubertal uterus that was 40 × 5 × 9 mm in size. Thyroid function test results were normal. A gonadotropin-releasing hormone (GnRH) agonist stimulation test yielded a basal LH level below 0.1 mIU/mL, with a peak level of 4.8 mIU/mL at 45 minutes. The test also yielded a basal FSH level of 8.5 mIU/mL, with a peak level of 49.9 mIU/mL at 90 minutes. The serum estradiol concentration was below
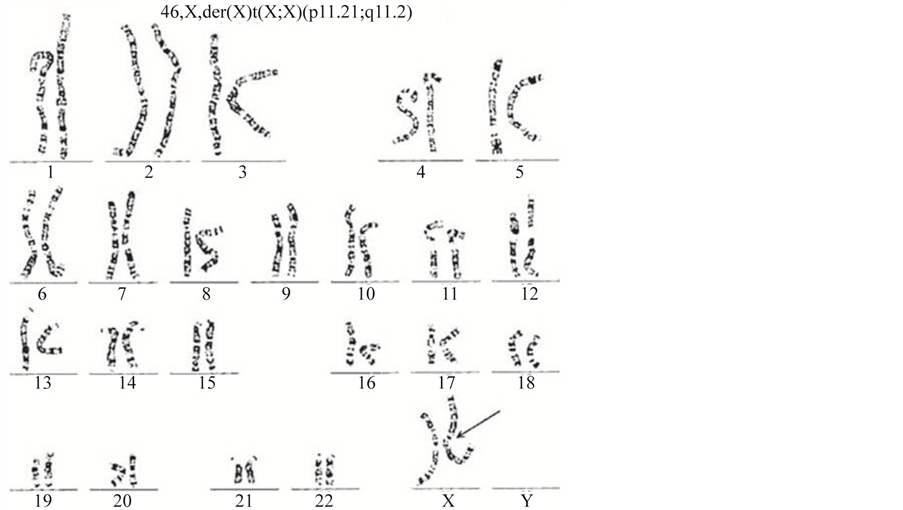
Figure 1. Karyotype results from our patient showing the 46, X, der (X) t (X; X) (p11.21; q11.2) pattern. The arrow indicates the deletion of the short arm and the duplication of the long arm.
5.0 pg/mL, and serum levels of IGF-1 and IGFBP-3 were within normal limits. Although these findings were not completely consistent with precocious puberty, some pubertal development was detected.
Growth hormone therapy was started to attempt to improve adult height by human recombinant somatotropin with the dose of 1 IU/Kg/week. Breast development did not progress and the breasts remained in the Tanner III stage. Bone age acceleration did not proceed. Her growth velocity improved; in the first year after GH treatment her growth rate was 7.2 cm/year, and it was 8.4 cm/year in the second year (Figure 2). She was 137.8 cm tall and weighed 45.5 kg at her last visit (Feb 2016).
3. Discussion
Rare derivative X chromosomes have long been investigated. The mechanism of their formation is of particular interest, as is the genotype-phenotype correlation in TS. Although the correlation between genotype and phenotype are not yet well understood [5] , genetic polymorphisms are believed to be associated with the clinical features of Turner syndrome [6] . A 45, X karyotype, which is the most frequent karyotype in Turner syndrome, arises more often by loss of the paternal X or Y chromosome in meiosis or in early embryogenesis than by loss of the maternal X chromosome [7] .
More than 20% of all patients with TS are diagnosed after 12 years of age [8] . Most patients with TS are thus

Figure 2. Growth curves of our patient.
detected in mid-childhood. By this time, most patients have failed to realize pubertal changes and have missed the window of opportunity for growth hormone therapy, resulting in significantly short stature.
Like complete monosomy X, partial deletions of either the short arm or long arm can cause features of TS. Many studies have been conducted to verify and delineate the proposed loci for genes related to the TS phenotype. Some studies have indicated that the genes for physical and cognitive features lie on Xp, whereas genes for ovarian function are present on both Xp and Xq (3). Ferguson-Smith, M.A. Karyotype-phenotype correlations in gonadal dysgenesis and their bearing on the pathogenesis of malformations. J Med Genet. 1965; 2: 142-155 [9] [10] .
Approximately one-third of all girls with TS enter spontaneous puberty, but only half of them complete with menarche [11] . The prevalence of spontaneous puberty is higher in patients with mosaic TS, although a few rare cases of TS have shown precocious puberty [11] - [16] .
In our case, karyotype analysis showed that a short arm was deleted from one of her X chromosomes and its long arm was duplicated. This phenomenon of short arm deletion/long arm duplication presents in approximately 18% of all TS cases. Short arm deletions from the X chromosome can result in short stature. Our case shows that patients with TS with a conserved long arm of the X chromosome can exhibit secondary sex characteristics. Our case highlights the possibility of natural pubertal development as an atypical clinical feature of TS and short stature as a typical clinical feature of TS. Precocious puberty in patients with TS is very rare; only five such patients have been reported, four of whom showed mosaic TS [13] [15] [16] . One of the patients had variant TS, as was found in our previously reported patient [14] . Precocious puberty may occur in girls with TS when cells with X chromosomes containing duplicated long arms undergo dosage compensation, thus allowing normal ovarian function.
Short stature is the single most common physical abnormality in TS. Four phases of growth have been established in TS: 1) moderate intrauterine growth retardation, 2) normal growth velocity between birth to 3 years of age, 3) decreasing growth velocity (height) between 3 to 12 years of age, and 4) failure to undergo a pubertal growth spurt [17] .
Our case did not have any TS-specific clinical hallmarks, with the exception of short stature, and her cognitive development was normal. She visited our clinic because of her pubertal development and short stature. Our case highlights the variability that can occur in patients with TS and emphasizes the need to carefully assess unusual growth patterns in any child, regardless of other underlying conditions.
Conflicts of Interest
No potential conflicts of interest relevant to this article are reported.
Cite this paper
Seok Ho Yoon,Dong Jun Lee,Son Moon Shin,So Young Yoon,Sung Won Park, (2016) A New Case of Turner Syndrome with Early Pubertal Development. International Journal of Clinical Medicine,07,342-346. doi: 10.4236/ijcm.2016.75036
References
- 1. Stochholm, K., Juul, S., Juel, K., Naeraa, R.W. and Gravholt, C.H. (2006) Prevalence, Incidence, Diagnostic Delay, and Mortality in Turner Syndrome. Journal of Clinical Endocrinology & Metabolism, 91, 3897-902.
http://dx.doi.org/10.1210/jc.2006-0558 - 2. Davenport, M.L. (2010) Approach to the Patient with Turner Syndrome. Journal of Clinical Endocrinology & Metabolism, 95, 1487-1495.
http://dx.doi.org/10.1210/jc.2009-0926 - 3. Donaldson, M.D., Gault, E.J., Tan, K.W. and Dunger, D.B. (2006) Optimising Management in Turner Syndrome: From Infancy to Adult Transfer. Archives of Disease in Childhood, 91, 513-520.
http://dx.doi.org/10.1136/adc.2003.035907 - 4. Bondy, C.A. (2007) Care of Girls and Women with Turner Syndrome: A Guideline of the Turner Syndrome Study Group. Journal of Clinical Endocrinology & Metabolism, 92, 10-25.
http://dx.doi.org/10.1210/jc.2006-1374 - 5. Ogata, T., Muroya, K., Matsuo, N., Shinohara, O., Yorifuji, T., Nishi, Y., et al. (2001) Turner Syndrome and Xp Deletions: Clinical and Molecular Studies in 47 Patients. Journal of Clinical Endocrinology & Metabolism, 86, 5498-5508.
http://dx.doi.org/10.1210/jcem.86.11.8058 - 6. Trovo de Marqui, A.B. (2015) [Turner Syndrome and Genetic Polymorphism: A Systematic Review]. Revista Paulista de Pediatria, 33, 364-371.
http://dx.doi.org/10.1016/j.rppede.2015.06.001 - 7. Hassold, T. and Hunt, P. (2001) To Err (Meiotically) Is Human: The Genesis of Human Aneuploidy. Nature Reviews Genetics, 2, 280-291.
http://dx.doi.org/10.1038/35066065 - 8. Massa, G., Verlinde, F., De Schepper, J., Thomas, M., Bourguignon, J.P., Craen, M., et al. (2005) Trends in Age at Diagnosis of Turner Syndrome. Archives of Disease in Childhood, 90, 267-268.
http://dx.doi.org/10.1136/adc.2004.049817 - 9. Ogata, T. and Matsuo, N. (1995) Turner Syndrome and Female Sex Chromosome Aberrations: Deduction of the Principal Factors Involved in the Development of Clinical Features. Human Genetics, 95, 607-629.
http://dx.doi.org/10.1007/BF00209476 - 10. Ross, J.L., Roeltgen, D., Kushner, H., Wei, F. and Zinn, A.R. (2000) The Turner Syndrome-Associated Neurocognitive Phenotype Maps to Distal Xp. American Journal of Human Genetics, 67, 672-681.
http://dx.doi.org/10.1086/303039 - 11. Improda, N., Rezzuto, M., Alfano, S., Parenti, G., Vajro, P., Pignata, C., et al. (2012) Precocious Puberty in Turner Syndrome: Report of a Case and Review of the Literature. Italian Journal of Pediatrics, 38, 54.
http://dx.doi.org/10.1186/1824-7288-38-54 - 12. Sandal, G. and Pirgon, O. (2014) Precocious Puberty in a Patient with Mosaic Turner Syndrome. Genetic Counseling, 25, 183-187.
- 13. Sabin, M.A. and Zacharin, M.R. (2007) Precocious Puberty in Turner Syndrome. Journal of Paediatrics and Child Health, 43, 776-778.
http://dx.doi.org/10.1111/j.1440-1754.2007.01219.x - 14. Baek, J.U., Park, H.K., Shim, E.J. and Hwang, I.T. (2012) Precocious Puberty in Turner Syndrome Variant. Journal of Pediatric & Adolescent Gynecology, 25, e113-e114.
http://dx.doi.org/10.1016/j.jpag.2012.05.017 - 15. Evanchec, K.A. and Rotenstein, D. (2005) Treatment of Precocious Puberty in Two Patients with Turner Mosaicism. Journal of Clinical Endocrinology & Metabolism, 18, 819-822.
http://dx.doi.org/10.1515/JPEM.2005.18.8.819 - 16. Huseman, C.A. (1983) Mosaic Turner Syndrome with Precocious Puberty. Journal of Pediatrics, 102, 892-894.
http://dx.doi.org/10.1016/S0022-3476(83)80021-3 - 17. Ranke, M.B., Pfluger, H., Rosendahl, W., Stubbe, P., Enders, H., Bierich, J.R., et al. (1983) Turner Syndrome: Spontaneous Growth in 150 Cases and Review of the Literature. European Journal of Pediatrics, 141, 81-88.
http://dx.doi.org/10.1007/BF00496795