Advances in Enzyme Research
Vol.2 No.2(2014), Article ID:47031,14 pages
DOI:10.4236/aer.2014.22011
Partial Purification and Characterization of the Rat Parotid Gland Protein Kinase Catalyzing Phosphorylation of Matured Destrin at Ser-2
Eriko Osumi1, Chihiro Kondo2, Mitsumasa Mizuno2, Takahiro Suzuki2, Mamoru Matsubara3, Kazuo Shimozato1, Takao Kanamori2*
1Department of Maxillofacial Surgery, School of Dentistry, Aichi-Gakuin University, Nagoya, Japan
2Department of Biochemistry, School of Dentistry, Aichi-Gakuin University, Nagoya, Japan
3Department of Bioscience and Biotechnology, Faculty of Bioenvironmental Science, Kyotogakuen University, Kyoto, Japan
Email: *takao@dpc.aichi-gakuin.ac.jp
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 4 May 2014; revised 8 June 2014; accepted 19 June 2014
Destrin, also called actin-depolymerizing factor (ADF), exists in resting parotid tissue as phosphorylated (inactive) and dephosphorylated (active) forms, and β-adrenergic stimulation of this tissue induces dephosphorylation of destrin. It is suggested that destrin dephosphorylation is involved in cortical F-actin disruption observed in parallel with β-agonist-induced amylase secretion. At present, the phosphorylation/dephosphorylation mechanism of destrin in parotid tissue is not known. We previously detected, in a crude rat parotid extract, a constitutively active protein kinase catalyzing phosphorylation of destrin; however, its identification has been hampered by difficulty in its enrichment. The purpose of this study was to explore a simple purification method(s) for this enzyme. To this end, we first developed a high-throughput dot-blot assay for the kinase with an anti-phosphodestrin antibody and then studied its purification by column chromatography on several media. We found that the kinase could be partially purified by sequential chromatography on DEAE-cellulose, phenyl-Sepharose, and hydroxyapatite columns. In each chromatography, however, the kinase could be eluted, at the cost of resolution, only by sharp increases in the elution power of the eluent; gradual increases in the elution power resulted in unacceptably poor recovery. We confirmed that enzymatic properties of the kinase were not basically altered during the purification. Further purification of the kinase was achieved by native polyacrylamide gel electrophoresis (PAGE), which resolved the kinase activity into two bands and separated the activity from most proteins (the kinase activity after PAGE was detected with destrin-coated polyvinylidene difluoride membranes and the anti-phosphodestrin antibody). The two bands seem to constitute the major destrin-phosphorylating activity in the resting rat parotid gland. We here report its partial purification and characterization together with the detection methods.
Keywords:Protein Kinase Purification, Protein Kinase Assay, Destrin Kinase, Actin-Depolymerizing Factor, Rat Parotid Gland

β-Adrenergic stimulation of the rat parotid gland induces potent α-amylase secretion through cAMP-dependent protein kinase activation [1] [2] . During the search for endogenous substrates of the kinase, we unexpectedly observed that two endogenous proteins are dephosphorylated in response to β-adrenergic stimulation of this tissue and identified them as destrin and cofilin [3] [4] . In the rat parotid gland, destrin is present more abundantly than cofilin [3] . The two proteins are structurally related actin-binding proteins [5] , and their dephosphorylated forms are active in binding to Fand G-actin and in depolymerizing and severing F-actin [5] . Destrin is also called actin-depolymerizing factor (ADF) [5] . In the resting parotid tissue, both destrin and cofilin are partly phosphorylated at Ser-2 of their matured forms (Ser-3 in each cDNA-derived sequence) [3] [4] . There exists a cortical actin layer below the plasma membrane of the parotid acinar cell, and it is thought that the layer serves as a barrier preventing contact of secretory granules with the luminal plasma membrane [6] . Because the cortical actin layer disappears upon β-adrenergic stimulation [6] , it seems possible that β-adrenergic stimulation-induced dephosphorylation of destrin and/or cofilin is involved in depolymerization of cortical F-actin and thereby in α-amylase secretion [3] .
In an attempt to better understand the phosphorylation/dephosphorylation mechanism on destrin and cofilin in the parotid gland, we previously searched for a constitutively active protein kinase catalyzing phosphorylation of destrin in this gland and detected an activity of such a protein kinase in the soluble fraction with an assay system utilizing native destrin as a substrate and employing western blotting with an antibody specifically recognizing destrin phosphorylated at Ser-2 [7] . We referred to the protein kinase as “destrin Ser-2 kinase” (DS2K), partially characterized the enzyme in a crude extract, and obtained results suggesting that DS2K is different from kinases (e.g. LIM-kinases 1 and 2 and testicular protein kinases 1 and 2) reported to phosphorylate destrin in other stimulated cells or tissues [7] . Toward the identification, we then started on its purification by chromatography on several media. However, the purification proved to be unexpectedly difficult mainly because of its poor recovery and irregular elution during chromatography. We then developed a high-throughput, high-sensitivity assay method employing dot-blotting procedure and continued our effort to purify the enzyme. After various attempts, we recently found that the enzyme activity could be recovered from chromatographic columns, at the cost of resolution, by drastic changes in elution conditions; although the purification efficiencies were limited, the enzyme activity was reproducibly recovered and could be further purified by native PAGE. Expecting these results to be useful for further study of the kinase, we here report partial purification and characterization of the kinase activity together with its detection methods.
2.1. Dot-Blot Assay for DS2K Activity
Destrin used as the substrate was prepared from rat stomachs as described previously [7] . Storage conditions of destrin were modified: the concentrated destrin preparation was mixed with an equal volume of ethylene glycol (final destrin concentration, 700 μg/ml), and stored at −30˚C. Destrin thus stored could be used for at least 10 months. The standard kinase assay mixture (total volume, 5 μl) contained 50 mM bis[2-hydroxyethyl]iminotris [hydroxymethyl]methane (Bis-Tris), pH 6.8, 5 mM magnesium acetate, 70 μg/ml destrin, 5% ethylene glycol (derived from the destrin preparation), 2 mM ATP, 3 mM 2-mercaptoethanol, and an enzyme preparation [7] . In preliminary experiments, we studied the effect of 5% ethylene glycol on DS2K activity and observed that it reduced the activity by about 10%. We, however, ignored this effect of ethylene glycol because it protected the destrin stock solution from freezing at −30˚C and allowed long storage of destrin. The reaction was initiated by the addition of ATP, carried out for 30 min at 30˚C, and terminated by the addition of 1.5 μl of a stop solution containing 20 mM Tris-HCl, pH 7.5, 80 mM EDTA, and 200 mM NaF. It was confirmed that when the stop solution was added to the reaction mixture containing an active enzyme preparation, destrin was not phosphorylated at all during 30 min of incubation at 30˚C. The mixtures lacking destrin served as the blank. Each sample then received 18.5 or 43.5 μl of a diluted stop solution (1.5 volumes of the stop solution was diluted with 5 volumes of water), and the resultant samples (usually 15 μl each) were dot-blotted onto a PVDF membrane (Immobilon-PSQ, Millipore) with the device previously reported [8] . Before blotting, the PVDF membrane was treated with methanol, rinsed in H2O, and equilibrated with the diluted stop solution. After all the samples were dot-blotted, the PVDF membrane was placed in a boiling water bath for 3 min and then immunostained with the anti-phosphodestrin antibody DS-Np, which was prepared by us and specifically recognizes matured destrin phosphorylated at Ser-2 [7] ; antibody-antigen complexes were visualized with alkaline phosphatase-conjugated goat anti-rabbit IgG (Promega) and Western BlueTM Stabilized Substrate for Alkaline Phosphatase (Promega) as described previously [7] except that the second antibody was diluted with Immuno Shot (Cosmo Bio). The staining was followed by visual inspection and terminated when the spots of interest reached desired intensities within a linear range for densitometry. The stained membranes were scanned with a model GT-7600S flat-bed scanner (Epson); spots of phosphorylated destrin were quantitated with the public domain software NIH Image 1.62, which was written by Wayne Rasband (National Institutes of Health, Bethesda, MD, USA), as described previously [7] .
2.2. Partial Purification of DS2K
The “100,000 × g supernatant fraction” was prepared from male Wistar rat parotid glands as described previously. The experimental procedures and care of animals were in accordance with “Regulation on Animal Experimentation at Aichi-Gakuin University School of Dentistry” and were approved by “the Experimental Animal Ethical Committee of Aichi-Gakuin University School of Dentistry” (Approval Numbers: AGUD 153 and 251). Briefly, male Wistar rats (about 200 g, fed ad libitum) were anesthetized with diethyl ether and killed by decapitation. Parotid glands, situated inferior and anterior to ears and embedded in subcutaneous adipose tissue, were collected, trimmed free of adhering tissue, and stored at −80˚C. The frozen tissue was thawed, minced, and homogenized in a glass-Teflon homogenizer containing 10 volumes of 20 mM Tris-HCl, pH 7.5, containing 0.28 M sucrose, 2 mM EDTA, 1 mM EGTA, 1 mM benzamidine, 0.2 mM phenylmethylsulphonyl fluoride, 10 μg/ml leupeptin, 12 μg/ml chymostatin, 2 μg/ml pepstatin A, and 0.1% 2-mercaptoethanol. The resultant homogenate was filtered through a single layer of gauze and centrifuged at 100,000 × g for 90 min; the 100,000 × g supernatant fraction was stored at −30˚C. The following procedure was carried out at 0˚C - 4˚C. The 100,000 × g supernatant fraction containing 12 mg of protein was applied to a DEAE-cellulose (DE53, Whatman) column (0.9 × 0.8 cm; bed volume, 0.5 ml) equilibrated with Solution A (20 mM Tris-HCl, pH 7.5, 1 mM benzamidine, 10 μg/ml chymostatin, 4 μg/ml leupeptin, and 0.1% 2-mercaptoethanol), and the effluent was discarded. The column was sequentially washed with 1.25 ml of Solution A, 15 ml of Solution A containing 40 mM NaCl, and 1.5 ml of Solution A containing 200 mM NaCl. The effluent of the third washing step with Solution A containing 200 mM NaCl was saved and mixed with 250 μl of 3.5 M (NH4)2SO4 [final (NH4)2SO4 concentration, 0.5 M]. The resultant sample was applied to a phenyl-Sepharose (Phenyl Sepharose High Performance, GE Healthcare) column (0.46 × 1.5 cm; bed volume, 0.25 ml) equilibrated with Solution B (20 mM Tris-HCl, pH 7.5, 0.5 mM benzamidine, and 0.1% 2-mercaptoethanol) containing 0.5 M (NH4)2SO4. The column was sequentially washed with 2 ml of Solution B containing 0.5 M (NH4)2SO4, 3 ml of Solution B containing 0.4 M (NH4)2SO4, and then 0.98 ml of Solution B containing 50% ethylene glycol. The effluent of the third washing step with Solution B containing 50% ethylene glycol was saved, mixed with 20 μl of 0.5 M sodium phosphate, pH 6.8 (final sodium phosphate concentration, 10 mM), and applied to a hydroxyapatite (Macro-Prep Ceramic Hydroxyapatite Type I 20 μm, Bio-Rad) column (0.46 × 0.6 cm; bed volume, 0.10 ml) equilibrated with Solution C (10 mM sodium phosphate, pH 6.8, 0.5 mM benzamidine, and 0.1% 2-mercaptoethanol). The column was washed with 0.4 ml of Solution C and then with 0.4 ml of Solution C containing additional 90 mM sodium phosphate, pH 6.8 (total sodium phosphate concentration in the eluent, 100 mM). The effluent of the second washing step was saved and dialyzed against 200 ml of 10 mM Tris-HCl, pH 7.5, containing 0.5 mM benzamidine and 0.1% 2-mercaptoethanol for at least 2 h with a device for dialysis [9] . As described under “Results”, the preparation thus obtained contained DS2K activity; it was stored at −4˚C and used as the “partially purified DS2K fraction” in later experiments. In some experiments, the partially purified DS2K fraction was concentrated to about 25 μl with Vivaspin 500 (5000 MWCO) (Sartorius). When larger quantities of the partially purified DS2K fraction were required, the volumes of the supernatant, the columns, and the eluents were proportionately increased.
2.3. Electrophoretic Procedures
Sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis (PAGE) was carried out as described previously [3] according to the method of Fling and Gregerson [10] with following modifications: separating gels (1 mm thick) contained 12% acrylamide; and gels were fixed in 45% methanol containing 9% acetic acid for 2 h, washed in water for 30 min, and stained for protein with ImperialTM Protein Stain (Thermo Fischer Scientific) according to manufacturer’s instruction. Some samples were concentrated by lyophilization. Native PAGE was carried out in 1-mm-thick, 5% acrylamide slab gels (N,N’-methylenebisacrylamide, 0.24%) with the same buffer system used for SDS-PAGE without addition of SDS. Samples for native PAGE contained 0.5% 2-mercaptoethanol. After native PAGE, gels were stained with ImperialTM Protein Stain or subjected to detection of DS2K activity.
2.4. Detection of DS2K Activity on Gels after Native PAGE
After native PAGE, the lane to which a DS2K-containing sample was applied was cut out and equilibrated twice for 5 min in 3 volumes of Solution D (50 mM Bis-Tris, pH 6.8, containing 5 mM magnesium acetate, 2 mM ATP, and 3 mM 2-mercaptoethanol). Before the treatment of the gel, a rectangle (typically, 5 mm × length of the separating gel) was drawn on a PVDF membrane (Immobilon-PSQ) with a pencil and cut out with at least 5-mm margins; the membrane was wetted with methanol, rinsed with water and then with 50 mM Tris-HCl, pH 7.5, and placed on three layers of filter paper wetted with 50 mM Tris-HCl, pH 7.5. After the excess buffer solution was absorbed with dry paper, 35 μg/ml destrin in 50 mM Tris-HCl, pH 7.5, was evenly applied to the rectangle on the PVDF membrane to make a final surface density of 84 μg/cm2. The PVDF membrane was blocked for 90 min with Tris-buffered saline (TBS) containing 1% bovine serum albumin (BSA) and 1% Tween-20, rinsed three times with 3 ml of 50 mM Bis-Tris, pH 6.8 (10 min/wash), and equilibrated with Solution D. The PVDF membrane with a destrin-coated rectangle was placed on 3 layers of filter paper wetted with Solution D; and on the rectangle, the equilibrated gel prepared above was placed. The whole sandwich was covered with Saran Wrap and incubated at 30˚C for 4 h (the gel was slightly pressed to the PVDF membrane with a weight), and then the membrane was treated for 3 min in a boiling water bath and subjected to immunostaining with the antiphosphodestrin antibody DS-Np as described above. The phosphodestrin immunostained on the membrane represented the DS2K activity in the gel.
2.5. Protein Assay
Protein concentrations were determined as in [11] (the total volume of the assay mixture was reduced), with BSA as a standard.
2.6. Statistical Analysis
Unless otherwise stated, data were presented as means ± SD of three independent experiments performed in triplicate. Statistical significance in difference between the mean of a sample and the hypothetical mean was analyzed by a two-tailed one-sample t-test with the program Prism 4 (GraphPad Software). Two-group comparison was performed by Student’s t-test with the same program. A P-value of 0.05 or less was considered statistically significant.
3.1. Verification of the Dot-Blot Assay for DS2K Activity
We previously introduced a specific assay method for DS2K activity [7] . To detect destrin phosphorylated during incubation, we employed western blotting, which was time-consuming and occasionally provided poor results derived from uneven transfer of proteins. We therefore replaced the western blotting by dot blotting. Figure 1(a) and Figure 1(b) show linear dependencies of destrin phosphorylation, detected by dot blotting, on the reaction time and on the enzyme quantity, respectively. The revised method thus worked well, allowed easy and rapid handling of more than 50 samples at a time, and also increased the assay sensitivity by at least 5-fold (data not shown). In the dot-blotting procedure, however, proteins in the sample are not separated as in western blotting; therefore, care was taken not to add too large amounts of enzyme preparations to the assay mixture.
3.2. Partial Purification of DS2K by Column Chromatography
The DS2K activity is essentially present in the 100,000 × g supernatant fraction of parotid tissue [7] . Figure 2(a) and Figure 2(d) show the elution profile of proteins and DS2K activity, respectively, on DEAE-cellulose chromatography. Under the conditions used, DS2K activity was retained by the column and eluted with the eluent containing 0.2 M NaCl after washing the column with a solution containing 40 mM NaCl. In preliminary experiments where DEAE-cellulose columns loaded with the supernatant fraction were washed under different conditions with increasing NaCl concentrations in eluents, we observed that elution of DS2K was not reproducible, and that the recovery was often poor (data not shown). After several attempts, we found that the reproducibility in DS2K elution is improved by sufficient washing of the columns, before elution of DS2K, with eluent containing low concentrations (e.g. 40 mM) of NaCl. The preliminary washing slowly eluted a large amount of amylase retained by the column; it is likely that elution of amylase as a concentrated band disturbs chromatography, partly elutes DS2K activity, and results in poor reproducibility in DS2K elution. We also noticed that the DS2K activity could efficiently be eluted, at the cost of resolution, by stepwise elution with a sharp increase in the concentration of NaCl (e.g. from 40 mM to 200 mM); slow gradual increases in NaCl concentrations in eluents resulted in poor recovery. It is likely that the activity of DS2K depends on some cellular component(s) weakly associated with the enzyme, and that simultaneous elution of such a component(s) with DS2K is required for efficient recovery of the enzyme activity. The experimental conditions for DEAE-cellulose chromatography, described above, were determined on the basis of these observations. Neither dephosphorylated nor phosphorylated destrin is retained by DEAE-cellulose columns under the conditions used [3] [7] ; endogenous phosphodestrin, which was present in the supernatant fraction and mainly contributed to the blank value in the assay used here (see the inset of Figure 1(a)), was eliminated by the DEAE-cellulose chromatography; the blank value of the fraction containing DS2K was thus essentially zero (data not shown).
We then searched for chromatomedia that could be used for further purification and reached a tentative conclusion that phenyl-Sepharose and hydroxyapatite could be candidates. In hydrophobic interaction chromatography

Figure 1. Detection of DS2K activity by dot blotting. (a) Time course. The rat parotid 100,000 × g supernatant (1 μg of protein) was incubated for the indicated times at 30˚C in the kinase assay mixture. The resultant reaction mixtures were processed for dot blotting as described under “Methods”. The inset shows a series of immunostained dots. The dot of the 0-min sample essentially represents the endogenous phosphodestrin present in the supernatant; the optical density of this dot was subtracted from the values of other dots, and the resultant values were used as the protein kinase activities; (b) Enzyme course. The rat parotid 100,000 × g supernatant, containing indicated amounts of protein, was incubated at 30˚C for 30 min in the kinase assay mixture. In each panel, values are means ± SD obtained in triplicate determinations; results are representative of three independent experiments.

Figure 2. Purification of DS2K by sequential chromatography on DEAE-cellulose, phenyl-Sepharose, and hydroxyapatite columns. (a), (d) The rat parotid 100,000 × g supernatant (12 mg of protein; 1.8 ml) was applied to a DEAE-cellulose column (0.9 × 0.8 cm). The flowthrough was collected as fraction 1 (lane 1). The column was sequentially washed with 1.25 ml of Solution A (lane 2), 3 × 5 ml of Solution A containing 40 mM NaCl (lanes 3 - 5), and 2 × 1.5 ml of Solution A containing 200 mM NaCl (lanes 6 and 7); the individual effluents were collected as fractions 2 - 7. Five percent of each fraction was dialyzed with a device for dialysis [9] ; resultant samples, together with the supernatant (Sup), were subjected to SDS-PAGE (a) and to DS2K activity assay (d). (b), (e) The volume of the remaining portion of fraction 6 from the DEAE-cellulose column was made up to 1.5 ml with Solution A containing 200 mM NaCl; to this solution, 250 μl of 3.5 M (NH4)2SO4 was added. The resultant sample was applied to a phenyl-Sepharose column (0.46 × 1.5 cm); the flowthrough was collected as fraction 1 (lane 1). The column was sequentially washed with 2 ml of Solution B containing 0.5 M (NH4)2SO4 (lane 2), 3 ml of Solution B containing 0.4 M (NH4)2SO4 (lane 3), and then 2 × 0.98 ml of Solution B containing 50% ethylene glycol (lanes 4 and 5); the individual effluents were collected as fractions 2 - 5. Five percent of each fraction was dialyzed before SDS-PAGE (b) and DS2K activity assay (e). (c), (f) The volume of the remaining portion of fraction 4 from the phenyl-Sepharose column was made up to 0.98 ml with Solution B containing 50% ethylene glycol; to this solution, 20 μl of 0.5 M sodium phosphate, pH 6.8, was added. The resultant sample was applied to a hydroxyapatite column (0.46 × 0.6 cm); the flowthrough was collected as fraction 1 (lane 1). The column was washed with 0.4 ml of Solution C (lane 2) and then with 2 × 0.4 ml of Solution C containing additional 90 mM sodium phosphate, pH 6.8 (lanes 3 and 4); the individual effluents were collected as fractions 2 - 4. Each fraction was dialyzed before SDS-PAGE (c) and DS2K activity assay (f). Panels (a)-(c) show areas from a single SDS-PAGE gel stained with ImperialTM Protein Stain; for each panel, portions collected from the dialyzed samples at a constant volume ratio were subjected to SDS-PAGE. Panels (d)-(f) show elution patterns of DS2K activity from the columns. Data represent means ± range obtained in duplicate determinations. In (a)-(c), the positions of standard proteins with the molecular masses indicated in kDa are shown on the right. Results are representative of three independent experiments. The arrow in (a) indicates the position of α-amylase.
on phenyl-Sepharose, samples containing high concentrations of NaCl can be directly applied to the columns. We therefore employed this chromatography in the second step. Figure 2(b) and Figure 2(e) show the elution profile of proteins and DS2K activity, respectively, on the phenyl-Sepharose chromatography. In the presence of 0.5 M (NH4)2SO4, DS2K was retained by the column and eluted by stepwise elution with a sharp increase in the elution power of the eluent (we used an eluent containing 50% ethylene glycol, which weakens the protein-ligand interactions). A slow gradual increase in the elution power of the eluent led to a poor recovery of the enzyme activity (data not shown). The active fraction was applied to a hydroxyapatite column, which was expected to retain DS2K in the presence of 50% ethylene glycol. Figures 2(c) and Figure 2(f) show the elution profile of proteins and DS2K activity, respectively, on the hydroxyapatite chromatography. DS2K was retained by the column, separated from ethylene glycol, and eluted by stepwise elution with an eluent containing 0.1 M sodium phosphate. A gradual increase in the concentration of sodium phosphate resulted in a low recovery (data not shown). A summary on representative purification of DS2K is shown in Table 1. In the repeated purification (n = 4) of DS2K by the procedure described above, we observed a total recovery of 3.6% ± 0.4% and a 2.3 ± 0.6-fold increase in the specific activity. Although the procedure separated DS2K from major abundant proteins, it resulted in a substantial loss of DS2K activity as well.
We also tried to use affinity chromatography on Aminophenyl-ATP-Sepharose (JENA Bioscience); when the active fraction from the DEAE-cellulose column was applied to the affinity column, DS2K activity was not retained by the column and eluted in the flowthrough fraction with essentially all other proteins (data not shown). This result might be due to the high Km (281 ± 22 μM) of DS2K for ATP [7] . We also attempted to use gel permeation chromatography on Sephacryl S-300 (GE Healthcare). Because the resolving power of this type of chromatography is limited, we have not obtained successful results.
3.3. Enzymatic Properties of DS2K Partially Purified by Column Chromatography
As described above, the enzyme activity was substantially lost during sequential chromatography on the three columns. We anticipated that the procedure led to a loss of some component(s) required for DS2K activity and to an alteration in the enzymatic properties. We therefore studied several key properties of DS2K in the partially purified fraction and compared them with previously reported properties of DS2K in the 100,000 × g supernatant fraction [7] . For DS2K partially purified in this work, the optimum pH was in the range 6.7 - 7.2 (Figure 3(a)); the Mg2+ concentrations required for maximum activity were 3 - 5 mM (Figure 3(b)); at 0.06, 0.1, 0.2, and 0.3 M, NaCl inhibited the activity by 56%, 70%, 84%, and 90%, respectively (Figure 3(c)). These properties of the partially purified DS2K were very similar to those of DS2K in the crude extract [7] . We then determined Km values of the partially purified DS2K for destrin and for ATP from Hanes-Woolf plots, where [S]/v was plotted against [S] ([S], substrate concentration; v, initial rate of reaction). The apparent Km values, each given by the negative of the [S]-axis intersection, for destrin and for ATP were determined to be 120 ± 20 μg/ml (n = 4) (Figure 3(d)) and 230 ± 40 μM (n = 4) (Figure 3(e)), respectively. When these values were compared with those previously obtained for DS2K in the crude extract [7] , they were not significantly altered (two-tailed unpaired t-test; P = 0.07 for Km toward destrin; P = 0.06 for Km toward ATP). From these results, we concluded that the enzymatic properties of DS2K were not essentially altered by its partial purification. As noticed previously [7] , the Km of DS2K for ATP was higher than the values (1 - 150 μM) reported for most other protein kinases [12] .
To characterize the DS2K activity in the crude extract, we previously studied the effect of several protein kinase inhibitors. We found that ML-7 (myosin light chain kinase inhibitor), W-7 (calmodulin inhibitor), Y-27632 (Rho-associated protein kinase inhibitor), and KN-93 (Ca2+/calmodulin-dependent protein kinase II inhibitor) were effective (e.g. ML-7 and W-7 inhibited the DS2K activity by more than 70% at 1 mM) [7] . In the present study, we again studied the effect of a series of protein kinase inhibitors on the partially purified DS2K activity to characterize this enzyme. The four inhibitors also suppressed the partially purified DS2K activity (Figure 3(f)).

Table 1. Purification of DS2K from rat parotid gland.
aValues were calculated for the case where samples were not collected for analysis from the column effluents.
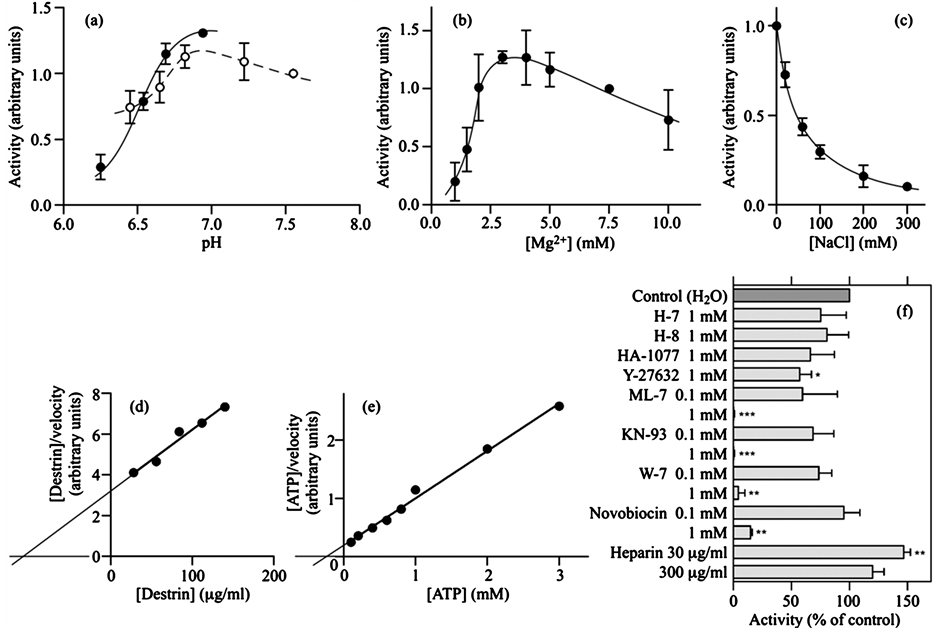
Figure 3. Enzymatic properties of DS2K in the partially purified DS2K fraction. (a) Effect of pH on DS2K activity. The pH of the kinase assay mixture was modified with 50 mM Bis-Tris (closed circles) and 50 mM MOPS (open circles) buffers. The activities were normalized by taking the activity at pH 7.55 as 1.0; (b) Effect of Mg2+ concentrations on DS2K activity. The activities were normalized by taking the activity at 7.5 mM Mg2+ as 1.0; (c) Effect of NaCl concentrations on DS2K activity. The activities were normalized by taking the activity at 0 mM NaCl as 1.0; (d) Km of DS2K for destrin. DS2K activity was determined in triplicate at indicated concentrations of destrin. The Hanes-Woolf plot of the results is shown. The Km for destrin, obtained from four experiments, was 120 ± 20 μg/ml (mean ± SD); (e) Km of DS2K for ATP. DS2K activity was determined in triplicate at indicated concentrations of ATP. The Hanes-Woolf plot of the results is shown. The Km for ATP, obtained from four experiments, was 230 ± 40 μM (mean ± SD); (f) Effects of protein kinase inhibitors on DS2K activity. DS2K activity was determined in the presence of each protein kinase inhibitor indicated on the left. All inhibitors were dissolved in H2O. The kinase activities thus obtained were normalized by taking the control value, which was obtained in the absence of inhibitors, as 100%. *P < 0.05; **P < 0.01; ***P < 0.001 (two-tailed one-sample t-test compared with 100). In (a)- (c) and (f), values are means ± SD of three experiments carried out in triplicate.
At 1 mM, ML-7 and KN-93 completely inhibited the activity; W-7 and Y-27632 suppressed the activity by 96% ± 6% (n = 3) and 43% ± 10% (n = 3), respectively. At 0.1 mM, the three inhibitors ML-7, KN-93 and W-7 did not significantly inhibit the activity (P = 0.14, P = 0.09, and P = 0.05, respectively). H-7 (a protein kinase C inhibitor), H-8 (cyclic nucleotide-dependent protein kinase inhibitor), and HA-1077 (a Rho-associated protein kinase inhibitor) did not effectively inhibit the partially purified DS2K activity (Figure 3(f)), as previously observed for the activity in the crude extract [7] . Thus, the relative potencies of protein kinase inhibitors also supported the notion that the properties of DS2K were not essentially altered. In the present study, we additionally found that novobiocin, an antagonist for heat shock protein 90 (Hsp90) and for bacterial gyrase B [13] , was also an effective inhibitor for DS2K activity; at 1 mM, it inhibited the DS2K activity by 85% ± 2% (n = 3) (Figure 3(f)). In our previous study, heparin (30 μg/ml), an inhibitor of protein kinase CK2 [14] , caused a statistically nonsignificant stimulation of DS2K activity in the crude extract [7] . In this study, heparin (30 μg/ml) significantly increased the partially purified DS2K activity by 47% ± 6% (P < 0.01, n = 3) (Figure 3(f)). We previously observed that staurosporine, a nonspecific protein kinase inhibitor, inhibited DS2K in the crude extract by 49% at 2 μM [7] ; however, it did not significantly inhibit the partially purified DS2K activity at 2 μM (P = 0.10, n = 3) (data not shown). Effects of protein kinase inhibitors would be further discussed below.
3.4. Purification of DS2K by Native PAGE
Further purification of DS2K by chromatography seemed difficult. We then attempted to purify DS2K by native PAGE because it might provide efficient resolution of DS2K and allow detection of the activity after electrophoresis. Since our previous study showed that unlike many other protein kinases, DS2K requires native destrin as the substrate and recognizes neither denatured destrin nor the peptide containing the phosphorylation site [7] , we introduced an on-gel detection method of DS2K activity after PAGE, utilizing destrin-coated PVDF membranes (see Figure 4). After native PAGE, a reference gel strip was cut out from a wide lane loaded with the partially purified DS2K fraction, equilibrated with a reaction mixture containing ATP, and placed on a destrin-coated PVDF membrane in a close contact with each other. After the stack was incubated at 30˚C for 4 h to allow DS2K to phosphorylate destrin on the membrane, the resultant PVDF membrane was subjected to immunostaining for phosphorylated destrin. A typical result is shown in Figure 4(a). DS2K activity was resolved into two bands each migrating slowly toward the anode during the native PAGE. It is likely that the two bands constitute the major activity phosphorylating destrin in the resting rat parotid gland. It remains to be clarified whether the two DS2K-derived bands represent isoforms of DS2K or two different enzymes. A gel strip was also cut from the gel adjacent to the reference gel strip, stained for protein with ImperialTM Protein Stain (Figure 4(b)), and aligned with the scanned image of the immunostained PVDF membrane; it was noticed that the DS2K-derived two bands were separated from most protein bands.
Because the DS2K-derived bands were well focused after native PAGE, it seemed possible that if the native PAGE gel was subsequently subjected to SDS-PAGE, the DS2K-derived bands would provide well-focused protein spots on the second dimension gel. We, therefore, subjected the native PAGE gel containing the two bands to SDS-PAGE, stained the resultant gel for protein with silver, and observed several spots under the two activity bands (see Figure S1). These spots were identified in separate experiments by proteomic analysis (see Figure S1 Legend); however, we have not detected protein kinase spots yet. It is likely that further enrichment of DS2K and/or improvement in mass spectrometry is required for identification of DS2K activity.
Identification of DS2K is expected to lead to better understanding on regulatory mechanism of destrin phosphorylation and of actin cytoskeleton in the parotid tissue. Proteomic approach for identification of DS2K could be facilitated by its preliminary enrichment; however, it has been hampered mainly by the poor recovery of DS2K

Figure 4. Native PAGE of DS2K. The partially purified DS2K fraction was prepared from the rat parotid 100,000 × g supernatant fraction containing 16 mg of protein, concentrated to about 50 μl with Vivaspin 500, and processed for native PAGE (the sample was applied onto a 50-mm-wide well of the stacking gel). Electrophoresis was carried out toward the anode. After PAGE, the separating gel under the wide well was cut out. At the center of the excised gel, a reference gel strip was cut longitudinally, leaving two gel strips with similar sizes. The central gel strip was processed for detection of DS2K activity with a destrin-coated PVDF membrane. The PVDF membrane immunostained for phosphodestrin is shown in (a); arrowheads indicate the positions of DS2K-derived kinase activities. The remaining two gel strips were stained with ImperialTM Protein Stain, and one of them is shown in (b) and aligned with the immunostained PVDF membrane (the scanned image of the PVDF membrane is enlarged to the size of the stained gel). Results are representative of three independent experiments. The gels stained for protein were further subjected to SDS-PAGE (see Figure S1).
in chromatography. In this study, we explored efficient procedure for enrichment of DS2K and found that sequential chromatography on DEAE-cellulose, phenyl-Sepharose, and hydroxyapatite could be used for partial purification of this enzyme. In each chromatography, however, effective recovery of DS2K required stepwise elution with a sharp increase in the elution power at the cost of the resolution; gradual increase in elution power resulted in poor recovery. A possibility therefore exists that DS2K exists as a loose complex with some cellular component(s), and that efficient recovery of DS2K activity requires its co-elution with such a component(s). We anticipated alteration of the enzymatic properties of DS2K during purification; however, properties of the partially purified DS2K were essentially similar to those of DS2K in the crude extract. It is likely that the recovered DS2K carries the required component(s).
The selectivity of protein kinase inhibitors has been extensively studied, and these studies have revealed that a number of so-called “specific” inhibitors affect so many protein kinases [15] -[17] . Among the protein kinase inhibitors studied in this work, ML-7, KN-93, W-7, and novobiocin effectively inhibited DS2K activity. The effects of ML-7 and KN-93, to say the least, seem to represent their nonspecific actions because it is reported that destrin is not effectively phosphorylated in vitro by myosin light chain kinase or by Ca2+/calmodulin-dependent protein kinase II [18] . The effect of W-7 and novobiocin might suggest the involvement of calmodulin and HSP90, respectively, in DS2K activity [13] [19] ; however, interpretation of these results requires caution. Some interaction of HSP90 with protein kinases has been reported [20] . The use of the four inhibitors as a set might help identification of DS2K. Requirement of high concentrations of each inhibitor is probably based on the facts that most protein kinase inhibitors are ATP-competitive [16] , and that our assay mixture contained a high concentration (2 mM) of ATP. Heparin increased the DS2K activity and might help identification of DS2K; however, the mechanism remains to be clarified.
In this work, we introduced a method for detection of DS2K activity on gels with destrin-coated PVDF membranes. In this method, gels after native PAGE were equilibrated with a reaction mixture containing ATP. We, however, observed that, proteins, including DS2K, partly flowed out from the gel during the equilibration. Although gels must be equilibrated before the subsequent kinase reaction, too long equilibration should be avoided. Upon on-gel assay of DS2K activity, we reproducibly detected two bands; however, band intensities were varied from one experiment to another. Further study is required to gain more reproducible results. It might be worth noting that the method introduced here allows detection of DS2K activity present only near the gel surface; in the case where DS2K is not sufficiently exposed to the gel surface after electrophoresis, its activity would be poorly detected.
In the 2D-PAGE employing native PAGE in the first dimension and SDS-PAGE in the second dimension (see Figure S1), it was expected that proteins moving as a complex during the first dimension would be separated during the second dimension SDS-PAGE and arranged vertically on the second dimension gel. Although we failed to find spots of DS2K and of possibly associated proteins after the 2D-PAGE, several points may deserve to note. Among the protein spots shown in Figure S1 and identified by proteomic analysis (see Figure S1 Legend), Sec13 and CSN4 are just below Sec31a and CSN2, respectively. It is reported that Sec13 and CSN4 are associated with Sec31 and CSN2, respectively [21] [22] ; Sec13 and Sec31 are components of the coat protein II complex (COPII) [21] [23] ; CSN2 and CSN4 are components of the COP9 signalosome (CSN) [22] . Thus, the 2D-PAGE used in this study probably allows detection of components of protein complexes. In addition, it was also noticed that deoxyribonuclease (DNase) I, which forms a complex with actin [24] , was also present below the actin spot (Figure S1).
In this work, we have attempted to purify and characterize DS2K, which is probably involved in phosphorylation of destrin in resting parotid tissue. Although additional work is required to better understand DS2K activity, we expect results reported in this paper to provide a basis for further study of the kinase and regulatory mechanism of actin cytoskeleton in the parotid gland.
This work was supported by grants-in-aid from the Aichi-Gakuin University.
1. Two-Dimensional PAGE (2D-PAGE)
After the first-dimension native PAGE, gels were stained with ImperialTM Protein Stain, destained in H2O, trimmed, equilibrated for 15 min with 62.5 mM Tris-HCl, pH 6.8, containing 0.3% SDS, 5% 2-mercaptoethanol and 10% glycerol, and loaded onto the second-dimension SDS-PAGE gels (0.8% agarose gel in the equilibration solution was used to keep the first-dimension gels in places); after the second-dimension electrophoresis, gels were stained with ImperialTM Protein Stain or with silver [1] .
2. Matrix-Assisted Laser Desorption/Ionization (MALDI) Time of Flight (TOF)-TOF Mass Spectrometry (MS) of Peptides
Sample preparation for mass spectrometry (in-gel reduction, alkylation, and trypsinization) was carried out according to the reported procedure [2] with the following modifications: proteins were reduced with 2 mM Tris(2-carboxyethyl)phosphine [3] [4] and alkylated with 140 mM acrylamide containing 140 mM 2-mercaptoethanol [3] [5] [6] . Briefly, protein spots were excised from gels stained with ImperialTM Protein Stain and cut into about 1 mm3 pieces; proteins were reduced, alkylated, and digested with 13 μg/ml trypsin (sequencing grade modified trypsin; Promega) overnight at 37˚C (usually, 10 μl of the trypsin solution was added to dried gel pieces); peptides generated were extracted from gels with 5% formic acid/acetonitrile (1:2, v/v), dried in a vacuum centrifuge, and dissolved in 0.1% trifluoroacetic acid.
A portion (0.5 μl) of each peptide solution prepared above was spotted on a sample area of an Opti-TOF 384-well plate. While the sample deposit was still wet, 0.5 μl of 10 mg/ml α-cyano-4-hydroxycinnamic acid (Sigma) in 50% acetonitrile/0.05% trifluoroacetic acid was added and allowed to dry. All mass spectra were obtained on a 4800 MALDI TOF-TOF Analyzer (Applied Biosystems) in the positive reflector mode; peptide mass fingerprint (PMF) data were first generated (scanning mass range, 700 - 4000 Da), and PMF data-dependent tandem MS (MS/MS) analysis was then performed (five most abundant precursors were selected for collision-induced dissociation MS/MS analysis; air was used as the collision gas). The processing of the data was carried out with the GPS Explorer software version 3.5 (Applied Biosystems), which submitted the data for a protein database search using an in-house licensed Mascot search engine (Matrix Science) with cysteine propionamidation and methionine oxidation as variable modifications (“Max. Missed Cleavage” was set at 1).

Figure S1. The gel shown in Figure 4(b) was cut at the positions corresponding to the kinase activity bands (arrowheads); the anodic portion of the gel was discarded. The resultant three gel pieces and the remaining whole gel strip (see Figure 4 and legend to it) were loaded on the second dimension gel for SDS-PAGE (some spaces were left between gels). A silver-stained gel after the second dimension SDS-PAGE is shown. Spots 1 - 7 were noticed because they were focused and present near or in contact with the empty lanes indicating the locations of the DS2K activity bands. The positions of standard proteins with the molecular masses indicated in kDa are shown on the right. Results are representative of three independent experiments. In separate experiments, we loaded twice the amount of the partially purified DS2K fraction onto 2D-PAGE, stained the gel after SDS-PAGE with ImperialTM Protein Stain, collected those corresponding to spots 1 - 7, and subjected them to reduction, alkylation, trypsin digestion, and MALDI-TOF-TOF MS followed by a database search. Spots 1 - 7 were identified as follows: spots 1 and 2, Sec31-like protein 1 (Sec31a; GI:55715648) (at least one of the two spots likely reflects post-translational modification of the protein [7] ); spot 3, Sec13-like 1 (Sec13; GI:54261633); spot 4, Nucleoporin 93 (GI:51859167); spot 5, Hsc70-ps1 (GI:56385); spot 6, COP9 constitutive photomorphogenic homolog subunit 2 (CSN2; GI:15215006); spot 7, COP9 signalosome subunit 4 (CSN4; GI:51259458). In addition, the two major spots with tailing were also identified: spot 8, β- and γ-actin (GI:55575 and GI:57573, respectively; we were unable to distinguish β-actin from γ-actin because they are different only in four residues located in their N-terminal regions); spot 9, DNase I (GI:15420826).
- Spearman, T.N. and Butcher, F.R. (1989) Cellular Regulation of Amylase Secretion by the Parotid Gland. In: Forte, J.G., Ed., Handbook of Physiology, Section 6, The Gastrointestinal System, American Physiological Society, Bethesda, 63-77.
- Takuma, T. and Ichida, T. (1994) Catalytic Subunit of Protein Kinase A Induces Amylase Release from Streptolysin O-Permeabilized Parotid Acini. The Journal of Biological Chemistry, 269, 22124-22128.
- Kanamori, T.,
Hayakawa, T., Suzuki, M. and Titani, K. (1995) Identification of Two 17-kDa Rat
Parotid Gland Phosphoproteins, Subjects for Dephosphorylation upon β-Adrenergic
Stimulation, as Destrinand Cofilin-Like Proteins. The Journal of Biological Chemistry,
270, 8061-8067.
http://dx.doi.org/10.1074/jbc.270.14.8061 - Kanamori, T., Suzuki, M. and Titani, K. (1998) Complete Amino Acid Sequences and Phosphorylation Sites, Determined by Edman Degradation and Mass Spectrometry, of Rat Parotid Destrinand Cofilin-Like Proteins. Archives of Oral Biology, 43, 955-967. http://dx.doi.org/10.1016/S0003-9969(98)00083-1
- Moon, A. and Drubin, D.G. (1995) The ADF/Cofilin Proteins: Stimulus-Responsive Modulators of Actin Dynamics. Molecular Biology of the Cell, 6, 1423-1431. http://dx.doi.org/10.1091/mbc.6.11.1423
- Perrin, D., Moller, K., Hanke, K. and Soling, H.D. (1992) cAMP and Ca2+-Mediated Secretion in Parotid Acinar Cells Is Associated with Reversible Changes in the Organization of the Cytoskeleton. The Journal of Cell Biology, 116, 127-134. http://dx.doi.org/10.1083/jcb.116.1.127
- Nakano, S., Kanamori, T., Suzuki, M. and Titani, K. (2003) Detection and Characterization of a Rat Parotid Gland Protein Kinase That Catalyzes Phosphorylation of Matured Destrin at Ser-2. Archives of Oral Biology, 48, 649-661. http://dx.doi.org/10.1016/S0003-9969(03)00129-8
- Kondo, C., Nakano, S., Suzuki, T. and Kanamori, T. (2007) An Easily Constructed, Inexpensive Device for Dot Blotting. Analytical Biochemistry, 370, 115-117. http://dx.doi.org/10.1016/j.ab.2007.05.015
- Armstrong, I.L. and Tate, W.P. (1980) A Simple Device for the Dialysis of Small Volumes. Analytical Biochemistry, 106, 469-470. http://dx.doi.org/10.1016/0003-2697(80)90549-7
- Fling, S.P. and Gregerson, D.S. (1986) Peptide and Protein Molecular Weight Determination by Electrophoresis Using a High-Molarity Tris Buffer System without Urea. Analytical Biochemistry, 155, 83-88. http://dx.doi.org/10.1016/0003-2697(86)90228-9
- Hartree, E.F. (1972) Determination of Protein: A Modification of the
Lowry Method That Gives a Linear Photometric Response. Analytical Biochemistry,
48, 422-427.
http://dx.doi.org/10.1016/0003-2697(72)90094-2 - Ruzzene, M. and Pinna, L.A. (1999) Assay of Protein Kinases and Phosphatases Using Specific Peptide Substrates. In: Hardie, D.G., Ed., Protein Phosphorylation, 2nd Edition, Oxford University Press, New York, 221-253.
- Marcu, M.G., Chadli, A., Bouhouche, I., Catelli, M. and Neckers, L.M. (2000) The Heat Shock Protein 90 Antagonist Novobiocin Interacts with a Previously Unrecognized ATP-Binding Domain in the Carboxyl Terminus of the Chaperone. The Journal of Biological Chemistry, 275, 37181-37186. http://dx.doi.org/10.1074/jbc.M003701200
- Hathaway, G.M., Lubben, T.H. and Traugh, J.A. (1980) Inhibition of Casein Kinase II by Heparin. The Journal of Biological Chemistry, 255, 8038-8041.
- Bain, J., McLauchlan, H., Elliott, M. and Cohen, P. (2003) The Specificities of Protein Kinase Inhibitors: An Update. Biochemical Journal, 371, 199-204. http://dx.doi.org/10.1042/BJ20021535
- Bain, J., Plater, L., Elliott, M., Shpiro, N., Hastie, C.J., McLauchlan, H., Klevernic, I., Arthur, J.S., Alessi, D.R. and Cohen, P. (2007) The Selectivity of Protein Kinase Inhibitors: A Further Update. Biochemical Journal, 408, 297-315. http://dx.doi.org/10.1042/BJ20070797
- Davies, S.P., Reddy, H., Caivano, M. and Cohen, P. (2000) Specificity and Mechanism of Action of Some Commonly Used Protein Kinase Inhibitors. Biochemical Journal, 351, 95-105. http://dx.doi.org/10.1042/0264-6021:3510095
- Morgan, T.E., Lockerbie, R.O., Minamide, L.S., Browning, M.D. and Bamburg, J.R. (1993) Isolation and Characterization of a Regulated Form of Actin Depolymerizing Factor. The Journal of Cell Biology, 122, 623-633. http://dx.doi.org/10.1083/jcb.122.3.623
- Hidaka, H. and Tanaka, T. (1983) Naphthalenesulfonamides as Calmodulin Antagonists. Methods in Enzymology, 102, 185-194. http://dx.doi.org/10.1016/S0076-6879(83)02019-4
- Caplan, A.J., Mandal, A.K. and Theodoraki, M.A. (2007) Molecular Chaperones and Protein Kinase Quality Control. Trends in Cell Biology, 17, 87-92. http://dx.doi.org/10.1016/j.tcb.2006.12.002
- D’Arcangelo, J.G., Stahmer, K.R. and Miller, E.A. (2013) Vesicle-Mediated Export from the ER: COPII Coat Function and Regulation. Biochimica et Biophysica Acta, 1833, 2464-2472. http://dx.doi.org/10.1016/j.bbamcr.2013.02.003
- Wei, N. and Deng, X.W. (2003) The COP9 Signalosome. Annual Review of Cell and Developmental Biology, 19, 261- 286. http://dx.doi.org/10.1146/annurev.cellbio.19.111301.112449
- Shugrue, C.A., Kolen, E.R., Peters, H., Czernik, A., Kaiser, C., Matovcik, L., Hubbard, A.L. and Gorelick, F. (1999) Identification of the Putative Mammalian Orthologue of Sec31P, a Component of the COPII Coat. Journal of Cell Science, 112, 4547-4556.
- Lazarides, E. and Lindberg, U. (1974) Actin Is the Naturally Occurring Inhibitor of Deoxyribonuclease I. Proceedings of the National Academy of Sciences of the United States of America, 71, 4742-4746. http://dx.doi.org/10.1073/pnas.71.12.4742
- Morrissey, J.H. (1981) Silver Stain for Proteins in Polyacrylamide Gels: A Modified Procedure with Enhanced Uniform Sensitivity. Analytical Biochemistry, 117, 307-310. http://dx.doi.org/10.1016/0003-2697(81)90783-1
- Shevchenko, A., Tomas, H., Havlis, J., Olsen, J.V. and Mann, M. (2006) In-Gel Digestion for Mass Spectrometric Characterization of Proteins and Proteomes. Nature Protocols, 1, 2856-2860. http://dx.doi.org/10.1038/nprot.2006.468
- Kanamori, T., Suzuki, M. and Titani, K. (1998) Complete Amino Acid Sequences and Phosphorylation Sites, Determined by Edman Degradation and Mass Spectrometry, of Rat Parotid Destrinand Cofilin-Like Proteins. Archives of Oral Biology, 43, 955-967. http://dx.doi.org/10.1016/S0003-9969(98)00083-1
- Wu, J., Gage, D.A. and Watson, J.T. (1996) A Strategy to Locate Cysteine Residues in Proteins by Specific Chemical Cleavage Followed by Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Analytical Biochemistry, 235, 161-174. http://dx.doi.org/10.1006/abio.1996.0108
- Brune, D.C. (1992) Alkylation of Cysteine with Acrylamide for Protein Sequence Analysis. Analytical Biochemistry, 207, 285-290. http://dx.doi.org/10.1016/0003-2697(92)90013-W
- Cavins, J.F. and Friedman, M. (1968) Specific Modification of Protein Sulfhydryl Groups with α, β-Unsaturated Compounds. The Journal of Biological Chemistry, 243, 3357-3360.
- Wei, N. and Deng, X.W. (2003) The COP9 Signalosome. Annual Review of Cell and Developmental Biology, 19, 261- 286. http://dx.doi.org/10.1146/annurev.cellbio.19.111301.112449

*Corresponding author.