Neuroscience & Medicine
Vol.5 No.2(2014), Article
ID:45542,12
pages
DOI:10.4236/nm.2014.52015
Nerve Growth Factor Enhances Tau Isoform Expression and Transcription in IMR32 Cells
Cheryl L. Cragg, Bettina E. Kalisch
Department of Biomedical Sciences, University of Guelph, Guelph, Canada
Email: bkalisch@uoguelph.ca
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 10 February 2014; revised 8 March 2014; accepted 7 April 2014
ABSTRACT
The present study characterized the nerve growth factor (NGF)-mediated regulation of tau protein expression and transcription in IMR32 human neuroblastoma cells. Treatment of IMR32 cells with 50 ng/mL NGF resulted in increased levels of specific tau protein isoforms. A 550 bp fragment of the tau promoter was cloned and treatment of transfected IMR32 and PC12 cells with NGF also resulted in increased promoter activation, suggesting that the NGF-mediated increase in tau isoforms is regulated, at least in part, at the level of transcription. Pretreatment with the MAP kinase inhibitor U0126 or the PKC inhibitor bisindolylmaleimide 1 (BIS-1) attenuated the NGF-mediated increase in tau transcription, indicating that the NGF-mediated activation of the MAP kinase and PKC signaling pathways modulate tau transcription. Pre-treatment of cells with the Akt inhibitor, LY294002 or with NOS inhibitors Nω-nitro-L-arginine methylester (L-NAME) or s-methylisothiourea (S-MIU) had no effect on the NGF-mediated increase in tau promoter activation, suggesting that NO and the NGF-Akt signaling pathway do not modulate tau transcription. Taken together, these data demonstrate that NGF increases the levels of multiple human tau isoforms in IMR32 cells which may result, at least in part, from NGF-mediated PKC and MAP kinase-induced tau transcription.
Keywords:Nerve Growth Factor (NGF), Tau, Signal Transduction, Transcription, Nitric Oxide (NO), IMR32 Cells, PC12 Cells

1. Introduction
The microtubule associated protein tau is found predominately in the axons of neurons and functions in microtubule assembly and stabilization, enabling axonal transport of cellular components back and forth from the cell body to the synapse [1] -[4] . Overexpression of tau protein impairs anterograde axonal transport of cellular components required for axon and synaptic maintenance [5] and hyperphosphorylation of tau results in the formation of paired helical filaments which aggregate to form neurofibrillary tangles that impair neuronal functioning and contribute to cell death [6] -[12] .
Axonal tau comes from a single gene, located on chromosome 17q21.1 in humans, which is alternatively spliced into 6 isoforms in the adult human brain [6] [13] . The tau gene produces 2, 6 and 8 kb transcripts which are differentially regulated depending on tissue type and developmental stage [6] [14] . The 2 kb transcript targets tau to the nucleus, the 9 kb transcript is specific to the retina and peripheral nervous system and the 6 kb transcript is translated into axonal tau isoforms [15] [16] . The six axonal isoforms are comprised of 352 to 441 amino acids which generate proteins that usually fall into the range from 45 to 67 kDa [17] [18] .
The axonal tau isoforms differ in two ways: 1) the carboxy-terminal presence of either 3 or 4 microtubule binding repeats (R), which are 31 to 32 amino acids long and 2) the absence (0N) or presence of either a 29 (1N) or 58 (2N) amino acid insert in the amino-terminus (N). These differences result in six tau isoform combinations that are referred to as 3R0N, 4R0N, 3R1N, 4R1N, 3R2N and 4R2N (reviewed in [19] ). In addition to its coding region, the tau gene has a 5’ exon designated-1 that is transcribed but not translated as well as 3’ untranslated regions which may be involved in the regulation of alternative splicing, subcellular localization and mRNA stability [20] . Splicing of tau RNA is complex and tightly regulated, with isoforms being expressed differently based on developmental stage and neuronal subpopulations [6] [18] . However upstream of exon-1, the tau promoter is TATA-less and has many CG rich regions, which is more characteristic of a housekeeping gene than one whose expression is tightly regulated [14] .
In addition to being differentially regulated during development, the tau gene can also be regulated by growth and transcription factors. Several studies have demonstrated that nerve growth factor (NGF) increases tau transcription in PC12 cells [17] [21] [22] . Heicklen-Klein et al. [23] demonstrated that the proximal portion of the rat tau promoter (−196 to +66) is activated by NGF in PC12 cells and that Sp1 and AP-2 transcription factors are responsible for the enhanced NGF responsiveness of this promoter fragment [23] [24] . Andreadis et al. [14] investigated a section of the human tau promoter upstream of exon-1 and found that the activation of this promoter was not different in Hela cells versus human neuroblastoma SKN cells, suggesting the neuronal specificity of the tau promoter was located outside of this promoter segment. The first objective of the present study was to evaluate the effect of NGF on human tau isoform expression and transcription.
Nitric oxide (NO) has been identified as an important modulator of NGF-mediated signaling and gene expression. In PC12 cells, NGF increases the expression of all three NO synthase (NOS) isozymes [25] -[27] and pretreatment with NOS inhibitors attenuates NGF mediated neurite outgrowth [25] [26] [28] [29] . We have also demonstrated that NO modulates NGF-mediated MAP kinase [30] and Akt [31] signaling, NGF-enhanced choline acetyltransferase activity and mRNA expression [25] and the NGF-induced expression of amyloid precursor protein [28] [32] and low density lipoprotein receptor-related protein 1 [33] . The second objective of the present study was to evaluate the role of NO and NGF signaling pathways in the NGF-mediated regulation of tau transcription.
Many studies looking to elucidate mechanisms involved in neuronal gene expression have been conducted in PC12 cells as these cells take on a cholinergic phenotype when differentiated with NGF [25] [34] . However, the IMR32 cell line has been identified as a good model for studying tau regulation as these cells have been shown to develop fibrillar structures that react to imunoprobes for paired helical filaments, the main constituents of neurofibrillary tangles [35] . In the present study, the NGF-mediated regulation of human tau protein expression was assesed in IMR32 cells and the transcriptional regulation of tau investigated in both IMR32 and PC12 cells.
2. Materials and Methods
2.1. Cell Culture and Treatments
IMR32 cells (ATCC, Manassas, VA, USA) were grown in Gibco® modified Eagle’s medium (MEM; Life Technologies, Burlington, ON, CAN) containing 10% fetal bovine serum (Hyclone, Logan, UT, USA), sodium pyruvate (1 mM) and 50 µg/mL gentamycin (Life Technologies). For protein analysis IMR32 cells were plated at a density of one million cells per 100 mm cellbind® plate (Corning, Lowell, MA, USA) the day before treatment. To establish a time course for NGF-mediated tau expression, IMR32 cells were left untreated (control) or treated with 50 ng/mL NGF (2.5 S Harlan Bioproducts, Indianapolis, IN, USA) and harvested at 24 h intervals for a maximum of 96 h. To evaluate the mechanisms involved in the NGF-mediated activation of tau, specific NGF signaling pathway or NOS inhibitors were added to the culture media 1 h prior to the addition of NGF (50 ng/mL). The inhibitor doses included 10 µM LY294002 (Calbiochem, San Diego, CA, USA; inhibits PI-3 kinase-mediated activation of Akt), 50 µM U0126 (Calbiochem; inhibits MEK1/2), the PLC-PKC inhibitors U-73122 and bisidolylmaleimide 1 (BIS-1; Calbiochem), 10 µM and 5 µM, respectively, 20 mM Nw-nitro-Larginine methylester (L-NAME; Sigma Aldrich, St. Louis, MO, USA; non-selective NOS inhibitor) and 2 mM s-methylisothiourea (S-MIU; Sigma; iNOS selective inhibitor). These inhibitors and doses were selected based on our previous studies on NGF-mediated signaling and gene expression in PC12 cells [25] [28] [30] -[33] . Media and treatments were replaced every 48 h.
2.2. Western Immunoblotting
Cells were rinsed with ice cold phosphate-buffered saline (PBS) followed by lysis in 250 µL of radioimmunoprecipitation assay (RIPA) buffer (final concentration: 50 mM Tris, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 0.5% sodium dodecylsulfate (SDS), 1 mM each of EDTA, sodium fluoride and sodium orthovanadate and one complete mini EDTA free tablet (Roche Diagnostics, Laval, QC, CAN)/10 mL of buffer). Samples were sonicated and the protein content of the supernatant determined by the method of Bradford [36] . Duplicate aliquots of some samples were dephosphorylated using 10 µL alkaline phosphatase (Roche Diagnostics) and 10 µL of the accompanying 10× dephosphorylation buffer (0.5 M Tris-HCl, 1 mM EDTA, pH 8.5), incubated at 37˚C for 60 min. Protein samples (100 µg) were boiled in SDS sample buffer with dithiothreitol (Cell Signaling Technology, Danvers, MA, USA) and a human tau protein ladder (Sigma Aldrich) was run alongside protein samples to identify tau isoform expression in the IMR32 cell line. Protein samples and standards were loaded onto 10% SDS/polyacrylamide separating gels and after electrophoresis, proteins were transferred onto nitrocellulose membranes using a semi-dry unit (Bio-Rad Laboratories, Mississauga, ON, CAN) with transfer buffer (48 mM Tris, 39 mM Glycine, 0.04% SDS and 20% methanol). After transfer, membranes were rinsed in tris-buffered saline (TBS; Tris 4 mM, NaCl 27.4 mM, pH 7.6) containing 0.1% Tween-20 (TBS-T) for 5 min then blocked for 1 h in TBS-T containing 5% skim milk and incubated overnight with mouse monoclonal tau antibody (Chemicon MAB3420; EMD Millipore, Billerica, MA, USA), 1:2500 in TBS-T with 5% skim milk. Blots were rinsed and incubated for 1 h with sheep anti-mouse IgG-horseradish peroxidase-conjugated secondary antibody (NA931; Amersham/GE Health Care, Piscataway, NJ, USA) 1:2000 in TBS-T with 5% skim milk followed by enhanced chemiluminescence (ECL; GE Healthcare). Protein bands were visualized using the STORM 860 scanner (Molecular Dynamics, subsidiary of Amersham) and densitometry was performed using Imagequant software (Molecular Dynamics). Following the detection of tau, blots were stripped and reprobed with mouse monoclonal α-tubulin antibody (1:50,000) in TBS-T containing 5% skim milk as described above.
2.3. Cloning of the Human Tau Promoter
IMR32 cells were plated on a 100 mm cell culture dish and grown to 90% confluency. Media was aspirated and cells were rinsed with cold PBS prior to isolating DNA with the DNeasy tissue kit (Qiagen, Mississauga, ON, CAN) according to the manufacturer’s directions. A 550 bp fragment of the tau promoter was amplified by polymerase chain reaction (PCR) using forward and reverse primers that included a Kpn 1 and Nhe 1 restriction site, respectively. Primer sequences were forward: 5’-CAGTGGTACCGACAATCTCTAGCTGG CCCTTAAAACATTC-3’ and reverse: 5’-CATAGCTAG CCTCCTGTAGTTGGAGTCTTTGTGTCG-3’. Purified PCR product (20 µL) and 6 µg of purified PGL3 basic vector (Promega Corporation, Madison, WI, USA) were digested with the restriction enzymes Kpn 1 and Nhe 1 (2 µL each; New England BioLabs, Ipswich, MA, USA) in NEBuffer 4 with BSA at 37˚C for 1 h. Following 20 min of heat inactivation at 65˚C samples were separated on a 1.5% agarose gel. The digested vector and PCR products were extracted from the gel and purified using the montage gel purification kit (EMD Millipore) and incubated with DNA ligase (Roche Diagnostics) at room temperature overnight. DNA from colonies of transformed DH5α cells (Life Technologies) was purified by alkaline lysis and the identity of the isolated DNA verified by restriction digest followed by DNA purification and sequencing (University of Guelph, Guelph Molecular Supercenter). DNA for transfections was purified using the Invitrogen Purelink HiPure Plasmid Filter purification kit (Life Technologies) according to the manufacturer’s directions.
2.4. Luciferase Assays
One day prior to transfection, IMR32 cells were plated onto 12 well dishes at a density of 300,000 cells per well in 2 mL media. Since the transfection efficiency and viability of the IMR32 cells was not as high as our previous experiments with PC12 cells, similar experiments were also conducted with PC12 cells. These cells were plated onto 12 well dishes at a density of 200,000 cells per well. On the day of transfection, media in each well was replaced with 1 mL antibiotic free media. Transfection solution containing 100 µL Opti-MEM medium (Life Technologies), 2 µL transfectin (Bio-Rad Laboratories), 1 µg tau promoter DNA and 0.5 µg β-galactosidase control vector DNA per well was prepared and incubated at room temperature for 30 min prior to its addition to the cell culture media. After 4 to 6 h of exposure to the transfection solution, the media was replaced with 2 mL of regular cell culture media.
The following day, cells were left untreated (control) or treated with NGF (50 ng/mL) or one of the inhibitors either alone or in combination with NGF. All treatments were administered to triplicate wells and inhibitors were added 1 h prior to NGF. Twenty-four h after NGF treatment cells were harvested in 200 µL 1× passive lysis buffer (Promega). Duplicate supernatant aliquots (20 µL for both luciferase and β-galactosidase analysis) were added to a 96-well plate. Prior to determining β-galactosidase activity, 25 µL Galacton, diluted 1:100 with reaction buffer diluent (Aurora Galacto-light kit, MP Biomedicals, Irvin, CA, USA) was added to each well and incubated for 30 to 60 min. The 96-well plates were read using a FLUOstar optima plate reader luminometer (BMG; Fisher Scientific, Ottawa, ON, CAN) which added 50 µL of the appropriate Accelerator reagent to each well immediately prior to luminescence quantification. For luciferase (Promega) the signal was read for 10 s following a 2 s delay and for β-galactosidase (Galacto-light) the signal was read for 1 s.
2.5. Data and Statistical Analysis
Following densitometric analysis with Image Quant, tau isoform protein levels were expressed relative to α-tubulin levels from the same sample. Luciferase values were normalized to β-galactosidase values from the same sample to control for transfection efficiency. All data are presented as the mean ± SEM and Graphpad Instat was used to analyze raw data. For most studies, sample size consisted of a minimum of 5 experiments. Statistical analysis was carried out by one-way analysis of variance (ANOVA) followed with Tukey-Kramer multiple comparisons post-hoc test. If data failed Bartlett’s test for comparable standard deviations or did not follow Gaussian distributions according to testing using the Kologorov and Smirnov method then data were assessed with the non-parametric ANOVA equivalent, the Kruskal-Wallace test, followed by Dunn’s multiple comparisons test.
3. Results
3.1. Expression of Tau Protein Isoforms in IMR32 Cells
A representative western blot depicting tau isoform expression in untreated (control) and NGF treated IMR32 cell lysates is shown in Figure 1. To identify the various tau isofoms, IMR32 cell extracts and dephosphorylated cell extract were compared to a tau protein ladder containing the six human tau isoforms (Sigma; data not shown). The smallest molecular weight isoform (0N3R) is the lowest band of the tau ladder and corresponds to the lowest molecular weight band detected in the upper panel of Figure 1. The other isoforms, in order from smallest to largest molecular weight, 0N4R, 1N3R, 1N4R, 2N3R, 2N4R appear above this band on the tau protein ladder and most were detected in the IMR32 cell lysates. As seen in Figure 1, the density of the tau bands detected in IMR32 cell lysates appeared to be greater for the 0N4R and 1N3R isoforms as compared to the other isoforms. The 0N3R and 1N4R isoforms were also detected consistently and could be reliably quantified. The 2N3R isoform was only detected in extracts obtained from IMR32 cells treated with NGF, indicating the expression of this isoform increased following NGF treatment, however this isoform was not detected consistently in all samples or on all blots and was therefore not quantified. The 2N4R isoform was rarely detected in cell extracts and was only observed on 50% of the blots analyzed. This isoform was also not quantifiable. Since dephosphorylated samples did not exhibit a different isoform band pattern from their unphoshorylated counterparts (not shown), all analysis of tau protein was completed using cell extract samples that were not dephosphorylated.
Densitometric analysis of tau protein revealed differential temporal regulation of tau isoforms following NGF treatment (Figure 2). Significant differences in the level of the 0N3R isoform were detected following NGF treatment (F(4, 55) = 3.979, p = 0.0066), with a significant increase observed following 72 h of NGF exposure

Figure 1. Tau isoform expression in control and NGF-treated IMR32 cells. Representative western blot obtained from IMR32 cell lysates separated by 10% SDS poly-acrylamide gel electrophoresis and transferred onto nitrocellulose membrane. The bands in the upper panel depict tau isoform levels and the lower panel shows α-tubulin protein expression from the same samples. The intensity and number of tau isoform bands appear to increase in response to increased duration of NGF exposure.
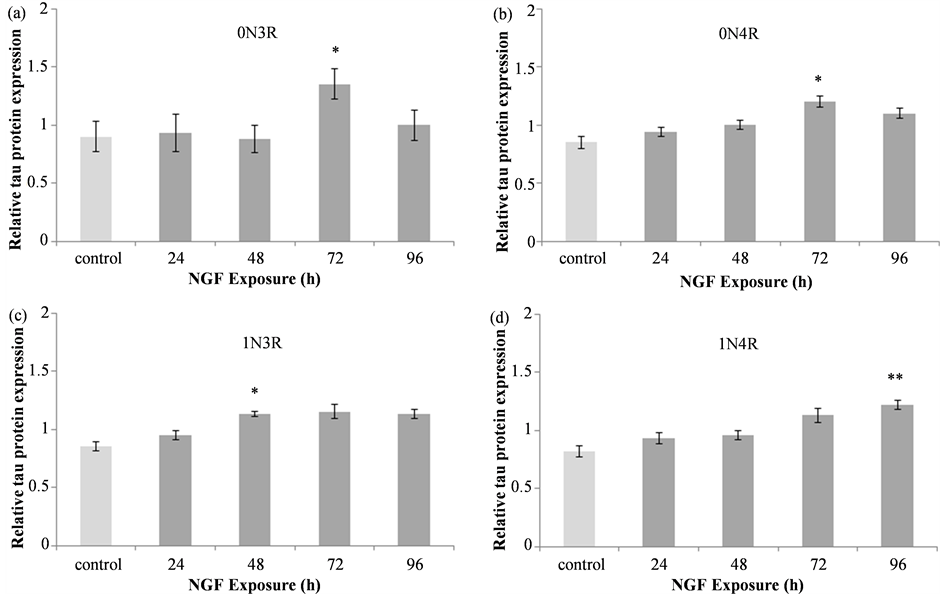
Figure 2. Tau isoform protein expression in NGF treated IMR32 cells. Summary of densitometric data from analysis of 0N3R (a), 0N4R (b), 1N3R (c) and 1N4R (d) levels relative to α-tubulin revealed a significant increase in tau isoform levels following treatment with 50 ng/mL NGF. Relative to control, a significant increase in 0N3R (*p < 0.05) and 0N4R (*p < 0.05) isoforms was detected following 72 h of NGF treatment. The levels of 1N3R were significantly higher than control after 48 h of NGF treatment (*p < 0.05) while 1N4R levels were elevated only after 96 h of NGF exposure (**p < 0.01).
as compared to control and all other treatment time points (Figure 2(a); *p < 0.05 vs. control and all other time points). The 0N4R isoform also exhibited significant differences in expression levels (F(4, 75) = 2.533, p = 0.0472), with increased levels detected at the 72 h treatment time compared to control (Figure 2(b)). Unlike the 0N3R isoform, the relative level of 0N4R detected following 72 h of NGF treatment was not significantly different from other time points suggesting that the level of this isoform at other treatment times falls somewhere between that observed in control and 72 h NGF treated lysates. The expression of the 1N3R isoform was also increased following NGF treatment (KW = 11.179, p = 0.0246). Although expression appeared increased at 72 h (Figure 2(c)) this difference was not quite statistically significant (p = 0.06) and the only statistically significant difference was in the 48 h NGF exposed cells, where the levels of this isoform were greater than control (*p < 0.05). The 1N4R tau isoform was also elevated following NGF treatment (F(4, 35) = 4.456, p = 0.0051) with significantly greater levels detected in cells treat with NGF for 96 h as compared to control (**p < 0.01; Figure 2(d)).
3.2. Tau Promoter Activation
To determine whether NGF affects the transcription of tau, a 550 bp fragment of the human tau promoter was cloned into the pGL3 basic luciferase reporter vector. Tau promoter activation in IMR32 and PC12 cells was determined by luciferase assay where luciferase activity was normalized to beta-galactosidase activity from the same sample.
In IMR32 cells (Figure 3(a)) NGF treatment did not increase luciferase activity in pGL3 basic-transfected cells relative to control cells but did increase luciferase activity in tau promoter-pGL3 basic transfected cells in comparison to control (t = 2.152, df = 14, p = 0.0494), indicating activation of the tau promoter by NGF. Since luciferase and beta-galactosidase activity was much lower than what we had detected with previous transfections using PC12 cells [28] [33] [37] tau promoter activation was also assessed in PC12 cells. In the PC12 cell line, both luciferase and β-galactosidase activities were much higher than those detected in IMR32 cells and a more robust NGF effect was observed (Figure 3(b)). One-way ANOVA revealed significant differences between treatment groups (F(5, 32) = 93.586, ***p < 0.001) and the Tukey-Kramer multiple comparisons test confirmed that NGF treatment significantly increased luciferase activity in cells transfected with the pGL3 basic vector alone (*p < 0.05) and in those transfected with the tau promoter construct (***p < 0.001). The NGF-mediated increase in luciferase activity in tau promoter-transfected cells was significantly higher than the pGL3-basic NGF response, indicating that NGF activates this fragment of the tau promoter. Based on these data, the PC12 cell line was used to assess the effects of NGF-TrkA signaling pathway and NOS inhibitors on the NGF-induced increase in tau promoter activation.
First, the effects of 50 µM UO126, which inhibits the MEK-MAP kinase pathway and 10 µM LY2940002, which inhibits the PI3K-Akt pathway were assessed (Figure 4(a)). There were no significant differences between the untreated control and inhibitor alone controls. NGF treatment increased promoter activation in both UO126 (*p < 0.001) and LY2940002 (***p < 0.001) pretreated cells in comparison to inhibitor alone controls. The NGF effect in UO126 pretreated cells was lower than that observed following treatment with NGF either alone (+p < 0.05) or pretreated with LY2940002 (p < 0.01) suggesting that the MAP kinase, but not Akt, signaling pathway is involved in the regulation of tau transcription.
The phospholipase C-PKC inhibitors U-73122 (10 µM) and BIS-1 (5 µM) were then used to investigate the involvement of the PKC signaling pathway. The effect of these inhibitors on NGF-induced tau promoter activation is shown in Figure 4(b). Statistical analysis revealed significant differences between treatments (F(5, 26) = 49.885, p < 0.0001). With both inhibitors, basal luciferase activity was not changed relative to control activity and for both control and inhibitor-pretreated cells, NGF resulted in a significant increase in relative luciferase activity (***p < 0.001). In comparison to treatment with NGF alone, in BIS-1 pretreated cells the NGF effect was significantly lower (+p < 0.05).
The effect of NOS inhibitors on human tau promoter activation is shown in Figure 4(c). Statistical analysis revealed significant differences between treatments (F(5, 28) = 96.971, p < 0.0001). In comparison to untreated cells, basal luciferase activity was not different in cells treated with 2 mM S-MIU or 20 mM L-NAME. In the presence or absence of NOS inhibitor, NGF treatment significantly increased luciferase activity relative to control (***p < 0.001), and there was no difference in the NGF response between control and NOS inhibitor pretreated samples.
4. Discussion
In the present study, it was shown for the first time that NGF is involved in the regulation of expression of multiple human tau isoforms in the IMR32 neuroblastoma cell line. This increased expression likely results, at least in part, from increased transcription as NGF also increased tau promoter activation in both IMR32 and PC12 cells. Inhibitors of MAP kinase and PKC activation attenuated this NGF mediated action, suggesting that the MAP kinase and PKC pathways play a role in regulating tau transcription. Neither LY294002 nor the NOS inhibitors, S-MIU or L-NAME, inhibited the NGF-mediated increase in tau promoter activation, suggesting that tau transcription is not modulated by the NGF-Akt signaling pathway or NO.
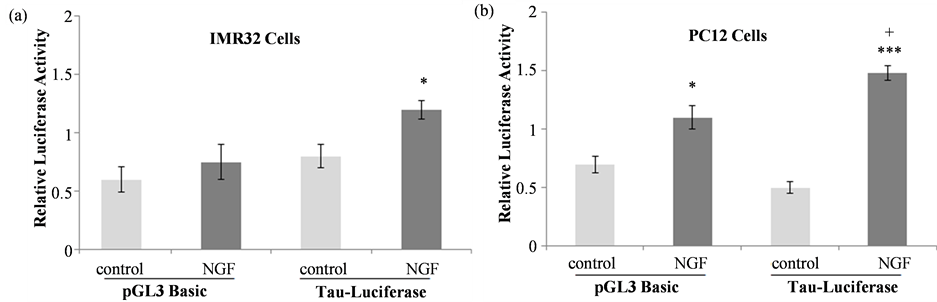
Figure 3. Tau promoter activation in response to NGF. IMR32 (a) or PC12 (b) cells transfected with the pGL3 basic vector or the tau promoter-pGL3 basic construct were treated with 50 ng/mL NGF. Luciferase activity in each sample was normalized to its corresponding β-galactosidase activity. NGF increased tau promoter activation in comparison to untreated controls in both cell lines, where * indicates significantly different activity from the corresponding control (*p < 0.05, ***p < 0.001) and + indicates significantly different NGF-mediated increase in luciferase activity compared to the pGL3 basic-NGF response (+p < 0.05).
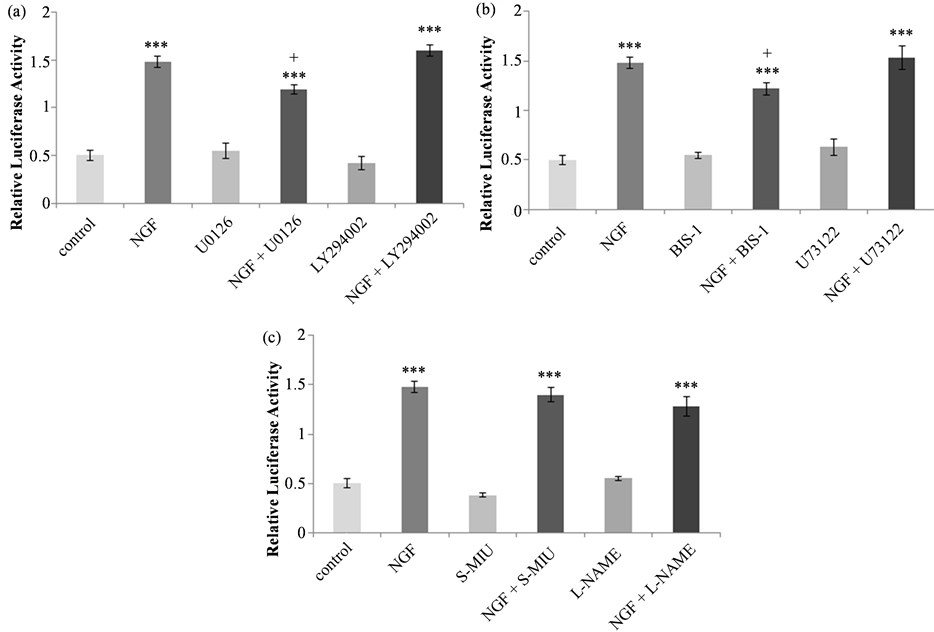
Figure 4. Tau promoter activation in PC12 cells in response to NGF in the presence or absence of signaling pathway or NOS inhibitors. (a) Effect of the inhibition of the MAP kinase pathway with 50 µM UO126 or the PI3 kinase-Akt pathway with 10 µM LY294002. NGF significantly increased luciferase activity in both untreated and inhibitor-treated cells (***p < 0.001). NGF-induced luciferase activity was significantly lower in cells pretreated with U0126, compared with NGF alone (+<0.05). (b) Effect of the inhibition of the phospholipase C-PKC pathway with 5 µM U73122 or 10 µM BIS-1. NGF significantly increased luciferase activity in in both untreated and inhibitor-treated cells (***p < 0.001). NGF-induced luciferase activity was significantly lower in cells pretreated with BIS-1, compared with NGF alone (+<0.05). (c) Effect of the inhibition of NOS with 2 mM S-MIU or 20 mM L-NAME. NGF significantly increased luciferase activity in both untreated and inhibitor-treated cells (***p < 0.001) and there was no difference in the NGF response in any of the treatment groups. For all inhibitors, there were no differences in luciferase activity in lysates of untreated controls or cells treated with inhibitor alone.
The 4 tau protein isoforms most prevalently expressed in human adult brain are those containing either no (0N) or one (1N) amino terminal insert, with both 3 and 4 repeat forms being expressed fairly equally [6] . The 4 main isoforms detected in the IMR32 cell line (0N3R, 0N4R, 1N3R, 1N4R) in this study correspond to the main isoforms expressed in adult human brain. We found that NGF increased the relative protein levels of all four of these isoforms in IMR32 cells. A significant increase in both the 3 and 4 repeat isoforms without an amino terminal insert (0N) was detected following 72 h of NGF treatment and increases in the levels of 1N3R were significantly higher after 48 h of NGF treatment while 1N4R levels were elevated only after 96 h of NGF exposure. A better understanding of the regulation of tau isoform expression, and in particular the larger 4R tau isoforms, is necessary as hyperphosphorylated 4R tau can result in reduced microtubule binding and tau accumulation and aggregation [38] . Although NGF was reported to induce tau phosphorylation in p75 expressing hippocampal neurons [39] , under ischemic conditions, NGF was reported to decrease hippocampal tau phosphorylation in rats [40] . As well, treatment of PC12 cells with NGF was reported to increase tau expression, without altering tau phosphorylation [13] . Since dephosphorylated samples did not exhibit a different isoform band pattern from their unphoshorylated counterparts, our findings are in agreement with those of Smith and colleagues [13] , and suggest that in IMR32 cells, NGF increases the expression of multiple tau isoforms via TrkA receptor activation without enhancing tau isoform phosphorylation.
To determine whether the NGF-induced increase in tau isoform expression was the result of altered transcription, a portion of the human tau promoter beginning approximately 200 bp upstream of the transcription start site and exon-1 and stretching roughly 550 bp upstream of this was cloned into the pGL3 basic luciferase reporter vector. Transfection of this construct revealed that this tau promoter segment was NGF responsive in both IMR32 and PC12 cells indicating that the NGF-mediated increase in tau isoform levels could be the result of enhanced tau transcription. Our findings with the human tau promoter are consistent with previous studies that have found the proximal portion of the rat tau promoter to be NGF responsive [3] [23] [24] . Heicklen-Klein et al. [23] demonstrated that the proximal 80 bp promoter portion of the rat tau promoter (−196 to +66) is responsive to NGF in PC12 cells. Andreadis et al. [14] compared tau exon-1 from the rat [17] to the human and found the sequences to be 91% conserved. We also observed greater promoter activation in the PC12 cell line compared to IMR32 cells. This enhanced transfection efficiency and promoter activation in PC12 cells was also reported by Heicklen-Klein and Ginzburg [24] , who transfected both COS (a non-neuronal cell line) and PC12 cells with a luciferase reporter containing the proximal portion of the rat tau promoter and found PC12 cells exhibited significantly greater promoter activation than that observed in the COS cells.
Since the NGF response was much more robust in the PC12 cells, this cell line was used to examine the effects of NOS and NGF-TrkA signaling pathway inhibitors on NGF-induced tau promoter activation. There was no significant difference in luciferase activity between the untreated control cells and cells treated with any of the inhibitors alone, suggesting that the inhibitors did not alter basal tau promoter activation. NGF significantly increased luciferase activity under all of the treatment conditions and none of the inhibitors tested completely blocked the NGF-induced activation of the tau promoter. The PKC inhibitor, BIS-1 attenuated the NGF-induced increase in luciferase activity while pretreatment with U73122 had no effect. U73122 inhibits phospholipase C and phospholipase C γ1 [41] -[45] , while BIS-1 shows high selectivity for inhibiting PKC-α, βI, βII, γ, δ, and ε isozymes [46] [47] . Our findings suggest that while activation of phospholipase C is not required for NGF-mediated tau promoter activation, activation of typical and/or novel PKC isoforms does contribute. Although PKC isoforms are activated by phospholipase C, they can also be activated by other factors that are stimulated by NGF. For example, NO is increased following treatment of PC12 cells with NGF and both NO donor drugs and peroxynitrite have been reported to activate PKC isoforms [48] [49] . NGF also stimulates the influx of calcium through membrane-bound ion channels [50] which could activate calcium-dependent PKC isoforms [51] . Therefore, inhibiting phospholipase C may not have prevented the NGF-mediated activation of certain PKC isoform that are necessary for tau promoter activation.
NGF-mediated promoter activation in UO126 pretreated cells was also significantly lower than that observed following treatment with NGF alone, suggesting that the MAP kinase pathway is involved in the regulation of tau transcription. We determined previously that NO modulates NGF-mediated MAP kinase activation [30] and the transcription of multiple genes that are upregulated by NGF [25] [28] [33] . Interestingly, NOS inhibitors had no effect on NGF-induced tau promoter activation in PC12 cells, suggesting that the regulation of tau transcription by MAP kinase is NO-independent. It has been reported that in PC12 cells, the MAP kinase pathway may act independently of other signaling pathways in the NGF-mediate regulation of gene expression [52] . Since U0126 and BIS-1 only partially inhibited NGF-stimulated tau promoter activation, it is possible that these NGFinduced pathways act independently to regulate tau transcription. There is also considerable evidence supporting cross-talk between these two pathways, in particular that PKC is required for the NGF-induced MAP kinase activation responsible for neurite outgrowth in PC12 cells [53] [54] . As well, both of these pathways are linked to a transcription factor that could be modulating tau transcription in our system. The human tau promoter fragment we cloned contains a GC rich region that could potentially be activated by the transcription factor Sp1. This transcription factor is increased following NGF treatment [55] and has been reported to modulate activation of the rat tau promoter [24] . Activation of PKC [56] or MAP kinase [57] was reported to regulate transcription at Sp1 binding sites and inhibition of MAP kinase using U0126 was found to decrease tumor necrosis factor α-induced Sp1 activation [58] . These findings suggest NGF-mediated activation of PKC and MAP kinase could enhance tau transcription through Sp1. Whether these signaling pathways act in a coordinated or independent manner to modulate NGF-induced tau transcription remains to be determined.
The link between the promoter activation, mRNA expression and resulting protein production can be very complicated since in addition to transcription and translation, the stability of the mRNA and protein could potentially play a significant role in the levels protein expressed in a cell. We have determined that NGF can regulate protein levels through both transcriptional and post-transcriptional mechanisms [28] [32] . Whether posttranscriptional mechanisms contribute to the NGF-mediated increase in tau isoform expression is not yet known. In addition, both NGF and Sp1 have been linked with the epigenetic mechanisms that could contribute to PC12 cell differentiation [59] [60] . In our system, it is possible that tau isoform expression is also modulated via NGFinduced epigenetic changes that modulate tau transcription.
Proper regulation of the expression and post-translational processing of tau is important for neuron function. The results from this study demonstrate that NGF increases the levels of multiple human tau isoforms in IMR32 cells and that this modulation may result, at least in part, from increased tau transcription. This modulation appears to involve the NGF-TrkA mediated activation of PKC and MAP kinase as inhibition each of these pathways partially prevented the NGF-mediated increase in human tau promoter activation.
Acknowledgements
These studies were supported by an operating grant from the Natural Sciences and Engineering Research Council of Canada to B.E.K. and an Ontario Graduate Scholarship to C.L.C.
References
- Binder, L.I., Frankfurter, A. and Rebhun, L.I. (1985) The Distribution of Tau in the Mammalian Central Nervous System. Journal of Cell Biology, 101, 1371-1378. http://dx.doi.org/10.1083/jcb.101.4.1371
- Cleveland, D.W., Hwo, S.Y. and Kirschner, M.W. (1977) Purification of Tau, a Microtubule-Associated Protein That Induces Assembly of Microtubules from Purified Tubulin. Journal of Molecular Biology, 116, 207-225. http://dx.doi.org/10.1016/0022-2836(77)90213-3
- Drubin, D.G., Feinstein, S.C., Shooter, E.M. and Kirschner, M.W. (1985) Nerve Growth Factor-Induced Neurite Outgrowth in PC12 Cells Involves the Coordinate Induction of Microtubule Assembly and Assembly-Promoting Factors. Journal of Cell Biology, 101, 1799-1807.
- Weingarten, M.D., Lockwood, A.H., Hwo, S.Y. and Kirschner, M.W. (1975) A Protein Factor Essential for Microtubule Assembly. Proceedings of the National Academy of Sciences of the United States of America, 72, 1858-1862. http://dx.doi.org/10.1073/pnas.72.5.1858
- Mandelkow, E.M., Stamer, K., Vogel, R., Thies, E. and Mandelkow, E. (2003) Clogging of Axons by Tau, Inhibition of Axonal Traffic and Starvation of Synapses. Neurobiology of Aging, 24, 1079-1085. http://dx.doi.org/10.1016/j.neurobiolaging.2003.04.007
- Andreadis, A. (2005) Tau Gene Alternative Splicing: Expression Patterns, Regulation and Modulation of Function in Normal Brain and Neurodegenerative Diseases. Biochimica et Biophysica Acta, 1739, 91-103. http://dx.doi.org/10.1016/j.bbadis.2004.08.010
- Billingsley, M.L. and Kincaid, R.L. (1997) Regulated Phosphorylation and Dephosphorylation of Tau Protein: Effects on Microtubule Interaction, Intracellular Trafficking and Neurodegeneration. Biochemical Journal, 323, 577-591.
- Kosik, K.S., Joachim, C.L. and Selkoe, D.J. (1986) Microtubule-Associated Protein Tau (Tau) Is a Major Antigenic Component of Paired Helical Filaments in Alzheimer Disease. Proceedings of the National Academy of Sciences of the United States of America, 83, 4044-4048. http://dx.doi.org/10.1073/pnas.83.11.4044
- Lovestone, S. and Reynolds, C.H. (1997) The Phosphorylation of Tau: A Critical Stage in Neurodevelopment and Neurodegenerative Processes. Neuroscience, 78, 309-324.
- Mandelkow, E.M., Biernat, J., Drewes, G., Gustke, N., Trinczek, B. and Mandelkow, E. (1995) Tau Domains, Phosphorylation, and Interactions with Microtubules. Neurobiology of Aging, 16, 355-362; discussion: 362-363.
- Ng, N.F. and Shooter, E.M. (1993) Activation of p21ras by Nerve Growth Factor in Embryonic Sensory Neurons and PC12 Cells. Journal of Biological Chemistry, 268, 25329-25333.
- Wood, J.G., Mirra, S.S., Pollock, N.J. and Binder, L.I. (1986) Neurofibrillary Tangles of Alzheimer Disease Share Antigenic Determinants with the Axonal Microtubule-Associated Protein Tau (Tau). Proceedings of the National Academy of Science United States of America, 83, 4040-4043. http://dx.doi.org/10.1073/pnas.83.11.4040
- Smith, C.J., Anderton, B.H., Davis, D.R. and Gallo, J.M. (1995) Tau Isoform Expression and Phosphorylation State during Differentiation of Cultured Neuronal Cells. Federation of European Biochemical Societies Letters, 375, 243-248. http://dx.doi.org/10.1016/0014-5793(95)01221-Y
- Andreadis, A., Wagner, B.K., Broderick, J.A. and Kosik, K.S. (1996) A Tau Promoter Region without Neuronal Specificity. Journal of Neurochemistry, 66, 2257-2263. http://dx.doi.org/10.1046/j.1471-4159.1996.66062257.x
- Nunez, J. and Fischer, I. (1997) Microtubule-Associated Proteins (MAPs) in the Peripheral Nervous System during Development and Regeneration. Journal of Molecular Neuroscience, 8, 207-222.http://dx.doi.org/10.1007/BF02736834
- Thurston, V.C., Pena, P., Pestell, R. and Binder, L.I. (1997) Nucleolar Localization of the Microtubule-Associated Protein Tau in Neuroblastomas Using Sense and Anti-Sense Transfection Strategies. Cell Motility and the Cytoskeleton, 38, 100-110. http://dx.doi.org/10.1002/(SICI)1097-0169(1997)38:1<100::AID-CM9>3.0.CO;2-C
- Sadot, E., Heicklen-Klein, A., Barg, J., Lazarovici, P. and Ginzburg, I. (1996) Identification of a Tau Promoter Region Mediating Tissue-Specific-Regulated Expression in PC12 Cells. Journal of Molecular Biology, 256, 805-812.http://dx.doi.org/10.1006/jmbi.1996.0126
- Sergeant, N., Delacourte, A. and Buée, L. (2005) Tau Protein as a Differential Biomarker of Tauopathies. Biochimica et Biophysica Acta (BBA), Molecular Basis of Disease, 1739, 179-197. http://dx.doi.org/10.1016/j.bbadis.2004.06.020
- Lee, G. and Leugers, C.G. (2012) Tau and Tauopathies. Progress in Molecular Biology and Translational Science, 107, 263-293. http://dx.doi.org/10.1016/B978-0-12-385883-2.00004-7
- Andreadis, A., Brown, W.M. and Kosik, K.S. (1992) Structure and Novel Exons of the Human Tau Gene. Biochemistry, 31, 10626-10633. http://dx.doi.org/10.1021/bi00158a027
- Drubin, D.G., Miller, K.G. and Botstein, D. (1988) Yeast Actin-Binding Proteins: Evidence for a Role in Morphogenesis. Journal of Cell Biology, 107, 2551-2561.
- Sadot, E., Barg, J., Rasouly, D., Lazarovici, P. and Ginzburg, I. (1995) Shortand Long-Term Mechanisms of Tau Regulation in PC12 Cells. Journal of Cellular Science, 108, 2857-2864.
- Heicklen-Klein, A., Aronov, S. and Ginzburg, I. (2000) Tau Promoter Activity in Neuronally Differentiated P19 Cells. Brain Research, 874, 1-9. http://dx.doi.org/10.1016/S0006-8993(00)02539-7
- Heicklen-Klein, A. and Ginzburg, I. (2000) Tau Promoter Confers Neuronal Specificity and Binds Sp1 and AP-2. Journal of Neurochemistry, 75, 1408-1418. http://dx.doi.org/10.1046/j.1471-4159.2000.0751408.x
- Kalisch, B.E., Bock, N.A., Davis, W.L. and Rylett, R.J. (2002) Inhibitors of Nitric Oxide Synthase Attenuate Nerve Growth Factor-Mediated Increases in Choline Acetyltransferase Expression in PC12 Cells. Journal of Neurochemistry, 81, 624-635. http://dx.doi.org/10.1046/j.1471-4159.2002.00854.x
- Peunova, N. and Enikolopov, G. (1995) Nitric Oxide Triggers a Switch to Growth Arrest during Differentiation of Neuronal Cells. Nature, 375, 68-73. http://dx.doi.org/10.1038/375068a0
- Sheehy, A.M., Phung, Y.T., Riemer, R.K. and Black, S.M. (1997) Growth Factor Induction of Nitric Oxide Synthase in Rat Pheochromocytoma Cells. Molecular Brain Research, 52, 71-77.http://dx.doi.org/10.1016/S0169-328X(97)00224-6
- Binnington, J.C. and Kalisch, B.E. (2007) Nitric Oxide Synthase Inhibitors Modulate Nerve Growth Factor-Mediated Regulation of Amyloid Precursor Protein Expression in PC12 Cells. Journal of Neurochemistry, 101, 422-433. http://dx.doi.org/10.1111/j.1471-4159.2006.04378.x
- Phung, Y.T., Bekker, J.M., Hallmark, O.G. and Black, S.M. (1999) Both Neuronal NO Synthase and Nitric Oxide Are Required for PC12 Cell Differentiation: A cGMP Independent Pathway. Molecular Brain Research, 64, 165-178. http://dx.doi.org/10.1016/S0169-328X(98)00315-5
- Kalisch, B.E., Demeris, C.S., Ishak, M. and Rylett, R.J. (2003) Modulation of Nerve Growth Factor-Induced Activation of MAP Kinase in PC12 Cells by Inhibitors of Nitric Oxide Synthase. Journal of Neurochemistry, 87, 1321-1332. http://dx.doi.org/10.1111/j.1471-4159.2003.02057.x
- Cheryl, L., Cragg, C.L., MacKinnon, J.C. and Kalisch, B.E. (2012) Nitric Oxide Synthase Inhibitors Modulate Nerve-Growth-Factor Mediated Activation of Akt. ISRN Cell Biology, 2012, Article ID: 847974.
- Mackinnon, J.C., Huether, P. and Kalisch, B.E. (2012) Effects of Nerve Growth Factor and Nitric Oxide Synthase Inhibitors on Amyloid Precursor Protein mRNA Levels and Protein Stability. Open Biochemistry Journal, 6, 31-39.http://dx.doi.org/10.2174/1874091X01206010031
- Grana, T.R., LaMarre, J. and Kalisch, B.E. (2013) Nerve Growth Factor-Mediated Regulation of Low Density Lipoprotein Receptor-Related Protein Promoter Activation. Cellular and Molecular Neurobiology, 33, 269-282.http://dx.doi.org/10.1007/s10571-012-9894-3
- Sandhu, F.A., Kim, Y., Lapan, K.A., Salim, M., Aliuddin, V. and Zain, S.B. (1996) Expression of the C Terminus of the Amyloid Precursor Protein Alters Growth Factor Responsiveness in Stably Transfected PC12 Cells. Proceedings of the National Academy of Sciences United States of America, 93, 2180-2185. http://dx.doi.org/10.1073/pnas.93.5.2180
- Ko, L.W., Sheu, K.F., Young, O., Thaler, H. and Blass, J.P. (1990) Expression in Cultured Human Neuroblastoma Cells of Epitopes Associated with Affected Neurons in Alzheimer’s Disease. American Journal of Pathology, 136, 867-879.
- Bradford, M.M. (1976) A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Analalytical Biochemistry, 72, 248-254.http://dx.doi.org/10.1016/0003-2697(76)90527-3
- Baskey, J.C., Kalisch, B.E., Davis, W.L., Meakin, S.O. and Rylett, R.J. (2002) PC12nnr5 Cells Expressing TrkA Receptors Undergo Morphological but Not Cholinergic Phenotypic Differentiation in Response to Nerve Growth Factor. Journal of Neurochemistry, 80, 501-511. http://dx.doi.org/10.1046/j.0022-3042.2001.00730.x
- Alonso, A.D., Zaidi, T., Novak, M., Barra, H.S., Grundke-Iqbal I. and Iqbal, K. (2001) Interaction of Tau Isoforms with Alzheimer’s Disease Abnormally Hyperphosphorylated Tau and in Vitro Phosphorylation into the Disease-Like Prtein. Journal of Biological Chemistry, 276, 37967-37973.
- Sáez, E.T., Pehar, M., Vargas, M.R., Barbeito, L. and Maccioni, R.B. (2006) Production of Nerve Growth Factor by β-Amyloid-Stimulated Astrocytes Induces p75NTR-Dependent Tau Hyperphosphorylation in Cultured Hippocampal Neurons. Journal of Neuroscience Research, 84, 1098-1106. http://dx.doi.org/10.1002/jnr.20996
- Zhang, Z.H., Xi, G.M., Li, W.C., Ling, H.Y., Qu, P. and Fang, X.B. (2012) Cyclic-AMP Response Element Binding Protein and Tau Are Involved in the Neuroprotective Mechanisms of Nerve Growth Factor during Focal Cerebral Ischemia/Reperfusion in Rats. Journal of Clinical Neuroscience, 17, 353-356.http://dx.doi.org/10.1016/j.jocn.2009.07.086
- Bleasdale, J.E., Thakur, N.R., Gremban, R.S., Bundy, G.L., Fitzpatrick, F.A., Smith, R.J. and Bunting, S. (1990) Selective Inhibition of Receptor-Coupled Phospholipase C-Dependent Processes in Human Platelets and Polymorphonuclear Neutrophils. Journal of Pharmacology and Experimental Therapeutics, 255, 756-768.
- Jones, N.P., Peak, J., Brader, S., Eccles, S.A. and Katan, M. (2005) PLCgamma1 Is Essential for Early Events in Integrin Signalling Required for Cell Motility. Journal of Cell Science, 118, 2695-2706.http://dx.doi.org/10.1242/jcs.02374
- Sato, K., Tokmakov, A.A., Iwasaki, T. and Fukami, Y. (2000) Tyrosine Kinase-Dependent Activation of Phospholipase Cgamma Is Required for Calcium Transient in Xenopus Egg Fertilization. Developmental Biology, 224, 453-469. http://dx.doi.org/10.1006/dbio.2000.9782
- Smith, R.J., Sam, L.M., Justen, J.M., Bundy, G.L., Bala, G.A. and Bleasdale, J.E. (1990) Receptor-Coupled Signal Transduction in Human Polymorphonuclear Neutrophils: Effects of a Novel Inhibitor of Phospholipase C-Dependent processes on Cell Responsiveness. Journal of Pharmacology and Experimental Therapeutics, 253, 688-697.
- Zapf-Colby, A., Eichhorn, J., Webster, N.J. and Olefsky, J.M. (1999) Inhibition of PLC-Gamma1 Activity Converts Nerve Growth Factor from an Anti-Mitogenic to a Mitogenic Signal in CHO Cells. Oncogene, 18, 4908-4919.http://dx.doi.org/10.1038/sj.onc.1202861
- Martiny-Baron, G., Kazanietz, M.G., Mischak, H., Blumberg, P.M., Kochs, G., Hug, H., Marmé, D. and Schächtele, C. (1993) Selective Inhibition of Protein Kinase C Isozymes by the Indolocarbazole Gö 6976. Journal of Biological Chemistry, 268, 9194-9197
- Toullec, D., Pianetti, P., Coste, H., Bellevergue, P., Grand-Perret, T., Ajakane, M., Baudet, V., Boissin, P., Boursier, E., Loriolle, F., Duhamelll, L., Charon, D. and Kirilovsky, J. (1991) The Bisindolylmaleimide GF 109203X Is a Potent and Selective Inhibitor of Protein Kinase C. Journal of Biological Chemistry, 266, 15771-15781.
- Bapat, S., Verkleij, A. and Post, J.A. (2001) Peroxynitrite Activates Mitogen-Activated Protein Kinase (MAPK) via a MEK-Independent Pathway: A Role for Protein Kinase C. Federation of European Biological Societies Letters, 499, 21-26. http://dx.doi.org/10.1016/S0014-5793(01)02511-X
- Liang, L.M. and Knox, F.G. (1999) Nitric Oxide Activates PKCalpha and Inhibits Na+-K+-ATPase in Opossum Kidney Cells. American Journal of Physiology, 277, F859-F865.
- Lichvárová, L., Jasková, K. and Lacinová, K. (2012) NGF-Induced Neurite Outgrowth in PC12 Cells Is Independent of Calcium Entry through L-Type Calcium Channels. General Physiology and Biophysics, 31, 473-478. http://dx.doi.org/10.4149/gpb_2012_054
- Dupont, J.L., Janoshazi, A., Bellahcene, M., Mykita, S. and De Barry, J. (2000) Reversible Protein Kinase C Activation in PC12 Cells: Effect of NGF Treatment. European Journal of Neuroscience, 12, 215-226.http://dx.doi.org/10.1046/j.1460-9568.2000.00906.x
- Park, S.Y., Lee, J.Y., Choi, J.Y., Park, M.J. and Kim, D.S. (2006) Nerve Growth Factor Activates Brain-Derived Neurotrophic Factor Promoter IV via Extracellular Signal-Regulated Protein Kinase 1/2 in PC12 Cells. Molecules and Cells, 21, 237-243.
- Corbit, K.C., Foster, D.A. and Rosner, M.R. (1999) Protein Kinase Cδ Mediates Neurogenic but Not Mitogenic Activation of Mitogen-Activated Protein Kinase in Neuronal Cells. Molecular and Cell Biology, 19, 4209-4218.
- Li, P., Yamakuni, T., Matsunaga, K., Kondo, S. and Ohizumi, Y. (2003) Nardosinone Enhances Nerve Growth Factor-Induced Neurite Outgrowth in a Mitogen-Activated Protein Kinaseand Protein Kinase C-dependent Manner in PC12D Cells. Journal of Pharmacological Science, 93, 122-125. http://dx.doi.org/10.1254/jphs.93.122
- Billon, N., Carlisi, D., Datto, M.B., van Grunsven, L.A., Watt, A., Wang, X.F. and Rudkin, B.B. (1999) Cooperation of Sp1 and p300 in the Induction of the CDK Inhibitor p21WAF1/CIP1 during NGF-Mediated Neuronal Differentiation. Oncogene, 18, 2872-2882. http://dx.doi.org/10.1038/sj.onc.1202712
- Park, K.M., Yule, D.I. and Bowers, W.J. (2009) Tumor Necrosis Factor-Alpha-Mediated Regulation of the Inositol 1, 4,5-Trisphosphate Receptor Promoter. Journal of Biological Chemistry, 284, 27557-27566.http://dx.doi.org/10.1074/jbc.M109.034504
- Benasciutti, E., Pagès, G., Kenzior, O., Folk, W., Blasi, F. and Crippa, M.P. (2004) MAPK and JNK Transduction Pathways Can Phosphorylate Sp1 to Activate the uPA Minimal Promoter Element and Endogenous Gene Transcription. Blood, 104, 256-262. http://dx.doi.org/10.1182/blood-2003-08-2661
- Lee, S.J., Park, S.S., Cho, Y.H., Park, K., Kim, E.J., Jung, K.H., Kim, S.K., Kim, W.J. and Moon, S.K. (2008) Activation of Matrix Metalloproteinase-9 by TNF-Alpha in Human Urinary Bladder Cancer HT1376 Cells: The Role of MAP Kinase Signaling Pathways. Oncology Reports, 19, 1007-1013.
- Bai, S., Ghoshal, K., Datta, J., Majumder, S., Yoon, S.O. and Jacob, S.T. (2005) DNA Methyltransferase 3b Regulates Nerve Growth Factor-Induced Differentiation of PC12 Cells by Recruiting Histone Deacetylase 2. Molecular and Cellular Biology, 25, 751-766. http://dx.doi.org/10.1128/MCB.25.2.751-766.2005
- Kato, A., Endo, T., Abiko, S., Ariga, H. and Matsumoto, K. (2008) Induction of Truncated Form of Tenascin-X (XB-S) through Dissociation of HDAC1 from SP-1/HDAC1 Complex in Response to Hypoxic Conditions. Experimental Cell Research, 314, 2661-2673. http://dx.doi.org/10.1016/j.yexcr.2008.05.019