Journal of Biophysical Chemistry
Vol.4 No.1(2013), Article ID:28478,6 pages DOI:10.4236/jbpc.2013.41003
Correlation of docking energies with spectroscopic kinetic assays of potential xanthine oxidase substrates
![]()
1Department of Pharmaceutical and Biomedical Sciences, Raabe College of Pharmacy, Ohio Northern University, Ada, USA; *Corresponding Author: a-stockert@onu.edu
2Pharmacy Administration, Ohio Health Grant Medical Center, Columbus, USA
Received 4 October 2012; revised 12 November 2012; accepted 20 November 2012
Keywords: Xanthine Oxidase; Computer Modeling; Docking; Inhibitor Design; Active Site Model
ABSTRACT
Here we present a docking model that ranks compounds according to their potential effectiveness as a potential substrate or inhibitor. We utilize xanthine oxidase (XO), a multi-cofactor oxido-reductase which converts hypoxanthine to xanthine and xanthine to uric acid. During the reductive half reaction, electrons flow from the molybdopterin, to each of two Fe/S centers, and finally to FAD. During the oxidative half reaction, electrons are passed from the FAD to O2. Under ideal physiological conditions, this reduction of oxygen generates H2O2 and, under multiple turnover conditions, superoxide in amounts which is regulated by catalase and superoxide dismutase. Utilizing computer modeling predictions of the docking orientations and energies of a group of purine based structures was selected. Correlating computer estimations with steady state kinetic data, a rapid screening process for inhibittor prediction was highlighted. This method allows educated selection of likely inhibitors, thereby decreasing the time and supplies required to complete a traditional kinetic analysis screening. Results demonstrate the functionality and reliability of this method and have proven particularly useful in understanding binding orienttations or poses of each compound.
1. INTRODUCTION
Docking is a method which predicts the preferred orientation of one molecule to a second molecule. It is a useful method of identifying different modes of binding for an inhibitor in an enzyme active site. By docking, one can identify which binding mode is more favorable and the results can be used to find inhibitors for target proteins and thus to design new drugs. Therefore, we use Glide® (Grid-base Ligand Docking with Energetics) program to generate ligand poses to search for possible locations of ligand molecules in the active-site region of the receptor. Our goal was to develop a more effective and high throughput method of ranking compounds according to potential function as inhibitors to the enzyme. We selected bovine xanthine oxidase based on its well characterized mechanism, available structures, and propensity to accept a wide variety of compounds in its active site. Additionally, the similarities between bovine xanthine oxidase and human xanthine oxidase make the bovine enzyme an appropriate medical target [1-9] .
Xanthine oxidase (XO) is an enzyme that converts hypoxanthine to xanthine and xanthine to uric acid, the last two steps in purine metabolism. XO is a molybdenum enzyme, which uses a unique mechanism of substrate hydroxylation. Unlike mono-oxygenase systems, the mechanism of molybdenum enzymes contains reductive and oxidative half reactions that are mediated by iron sulfur centers and uses oxygen gas as its final electron acceptor; other enzymes such as cytochrome oxidase c in the electron transport chain use O2 which is then reduced to H2O allowing the reaction to move forward [4].
Because O2 is the final electron receptor, XO generates reducing equivalents as a product of reaction. These reducing equivalents do not pose a threat under normal conditions because they are released at a rate that can be easily broken down by enzymes such as catalase and superoxide dismutase. However, during an ischemic condition such as a stroke or a heart attack, the oxidative half reaction is inhibited, leading to a stockpile of reducing equivalents, particularly on the flavin coenzyme. These reducing equivalents remain in the enzyme until oxygen is available to accept the electrons and restart the oxidative half reaction. Upon reintroduction of oxygen, these reducing equivalents rapidly convert oxygen to H2O2 and 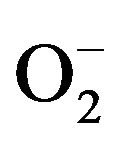 at a rate that cannot be broken down by other enzymes, leading to oxidative tissue damage, the primary cause of post myocardial infarction resulting in the decline of heart function [10-18] .
at a rate that cannot be broken down by other enzymes, leading to oxidative tissue damage, the primary cause of post myocardial infarction resulting in the decline of heart function [10-18] .
Allopurinol, a common XO inhibitor, is most recognized for its use in the treatment of gout where its efficacy is substantial [19]. Despite the success of allopurinol, exploration for new inhibitors is necessary due to potentially severe hypersensitivity and intolerance for patients with renal failure [20]. Additionally, allopurinol has been shown to reduce the severity of oxidative damage following ischemia; however, this helps only in patients already on allopurinol therapy [18,21-32] . Allopurinol originally binds to the active site of XO and is converted to alloxanthine and then must rebind in the active site to exert its inhibitory action. The nitrogen in position 8 prevents the carboxylic acid from forming at that position and also stops the flow of electrons to oxygen and therefore the number of reactive equivalents produced will be reduced [4].
Through the recent advent of computer modeling technology various compounds can be tested, their binding energies estimated, and their inhibitory potential evaluated. This will reduce the timely and costly kinetic studies required to screen any potential number of inhibitors which do not bind favorably. This particular study was conducted in order to verify that the computer model could provide a positive correlation between the two methods of kinetic spectroscopy and computer docking. The hypothesis tested was that Glide® docking energies would correlate with spectroscopic kinetic assays conducted with xanthine oxidase and potential substrates [33]. This method was also utilized to identify important contacts between the enzyme and the inhibitor, which may aid in the design of more effective inhibitors.
2. MATERIALS AND METHODS
2.1. Kinetic Analysis
Xanthine oxidase was purified from cow’s milk using ammonium sulfate precipitation, butanol extraction and size exclusion chromatogaraphy as previously described [34]. Enzyme concentration was measured by examining the absorbance at 450 nm and calculated using the extinction coefficient 37.8 mM/cm AU. Enzyme activity was estimated by measuring the uric acid production at 295 nm upon addition of xanthine and ranged from 37% to 57%. Final reaction concentration was corrected based on the percent of active enzyme. Reactions were run in triplicate with varying concentrations of compounds 1H -benzotriazole, 1H-benzotraizole-1-methanol, 1H-benzotraizole-5-carboxylic acid. Kinetic data was collected over a time interval of 4 minutes using and Hewlett-Packard 8424A spectrophotometer and a regression analysis using Chemstation biochemical software was preformed to provide and initial kinetic rate. Initial rates were averaged and plotted in a Lineweaver-Burke Plot allowing estimation of KI. The kinetic values were correlated to docking energies (using an ordinal classification), which take into account electrostatic and hydrophobic forces only, and therefore do not represent covalent binding energy. KI is the most appropriate parameter for comparison to docking energies for potential inhibitors.
2.2. Model Development
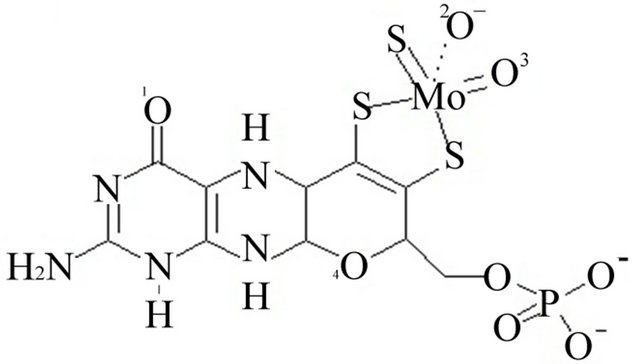
The coordinates file for bovine xanthine oxidase (PDB code 1FIQ) [35] was downloaded from the protein data bank (www.pdb.org). It could not be determined whether oxygen number two of the molybdopterin cofactor is ionized or neutral, Figure 1. The iron in the iron sulfur clusters was treated at Fe2+. The iron sulfur clusters in this enzyme are far from the active site where inhibitors were docked and, therefore, Fe2+ does not interact with the inhibitors. Therefore there are no contributions from Fe2+ to the docking energy estimated results will not be affected by the iron. Therefore, two copies of the coordinates file were prepared. In the first (ionized), the proton attached to oxygen two of molybdopterin was removed and a negative charge was added on that oxygen, Figure 1 panel A. In the second (neutral), the proton was not removed and the oxygen was left neutral, Figure 1 panel B. All bonds from protein atoms to the metal cofactors (the iron sulfur clusters and molybdenum (MO)) were deleted and hydrogen was added to all the atoms.
 (a)
(a)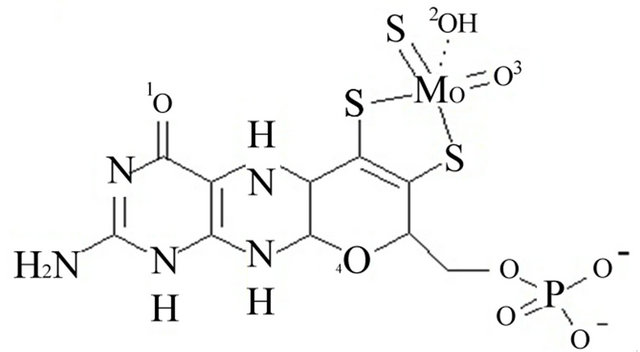 (b)
(b)
Figure 1. Panel A the ionized form of oxygen number 2. Panel B the neutralized oxygen number 2.
2.3. Docking
Glide® was used to calculate binding energies and orientations on 1H-benzotriazole, 1H-benzotriazole-1-methanol, and 1H-benzotriazole-5-carboxylic acid [33]. A grid box of 20 Å dimension in each direction was built around the active site centered on MO. A smaller region of 12 Å in each direction within this box was defined for inhibitor docking. Docking was performed with Glide® in the extra precision mode (XP) with the ligand poses per docking run was set to 2000 and the number of poses to be generated per ligand was set to 5. For each compound docked, any docked conformation with an energy value of more than 3 kCal/mol was discarded and only the five lowest energy poses were saved. The Root Mean Square (RMS) deviation was set to 1.0 Å and the maximum atomic displacement was set to 1.3 Å to insure that each of the generated five poses is significantly different from the other. All other docking parameters were left to their default values.
3. RESULTS
To test if the active site was represented correctly and to validate our docking protocol, a potent known inhibitor that has been crystallized with xanthine oxidase (TEI; PDB code 1N5X) [36,37] was docked into our model as prepared. Figure 2 shows that the docked conformation of TEI to be almost identical to its conformation in the crystal indicating the validity of our docking model and protocol.
Three substrates (1H-benzotriazole, 1H-benzotriazole-1-methanol, and 1H-benzotriazole-5-carboxylic acid) were analyzed kinetically in addition to being docked in the model active site their docking energies along
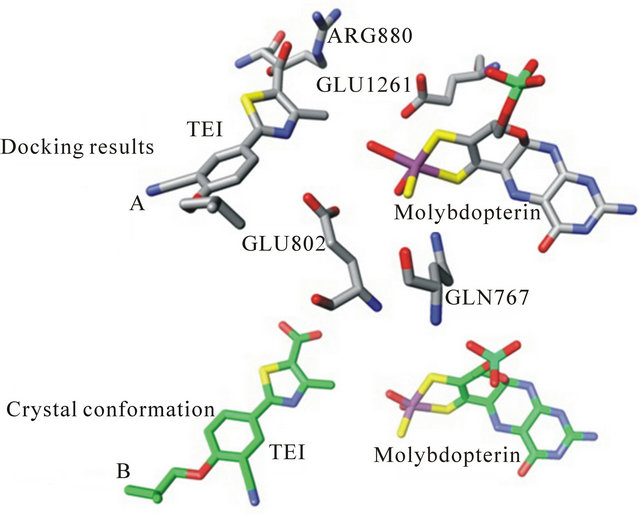
Figure 2. The crystal structure conformation of bovine xanthine oxidase with the inhibitor TEI bound (panel A, bottom). Panel B represents our docking model with the inhibitor TEI docking pose.
with their KI are shown in Table 1. Docking energies were selected from multiple binding poses selecting the lowest energy values. The calculated KI, based on kinetic data, is shown for each compound. The correlation between docking energies and KI support the use of this computer model to screen for potential inhibitors of this enzyme system. Compounds with predicted greater specificity result in greater affinity and lower binding energies. The prediction made about the KI based on the computer model came from individual poses which were taken and then analyzed in terms of functional groups relative to the position in the active site. The docking energies were then obtained compared experimental kinetic data. Figure 3 shows the docking pose of 1Hbenzotriazole-5-carboxylic acid in the ionized and neutral forms. In the neutral form, a hydrogen bond is preferred between the carboxylic acid group and the molybdopterin oxygen thus allowing the substituted group to have a greater interaction with the active moiety. In the ionized form, the hydrogen bond is formed between triazole nitrogen and the molybdopterin oxygen, which changed the orientation of the inhibitor so that it was opposite the neutralized form in the active site. Additionally, the phenyalanines in the active site adi to orient the substrate ring horizontal relative to the phenylalanines. Electrostatic interactions exist between the negative electron density on the triazole nitrogen and arginine 880. The differences in the interaction between the ionized and the neutral carboxylic acid group also accounts for the ionized group having a slower turnover and therefore a lower KI. Kinetic analyses reveal a KI of 12.8 mM for 1H-benzotriazole-5-carboxylic acid determined from the x-intercept of a secondary plot of the slopes from the Lineweaver-Burk plot.
1H-benzotriazole-1-methanol docking orientation is shown in Figure 3. Computer estimations predicted two
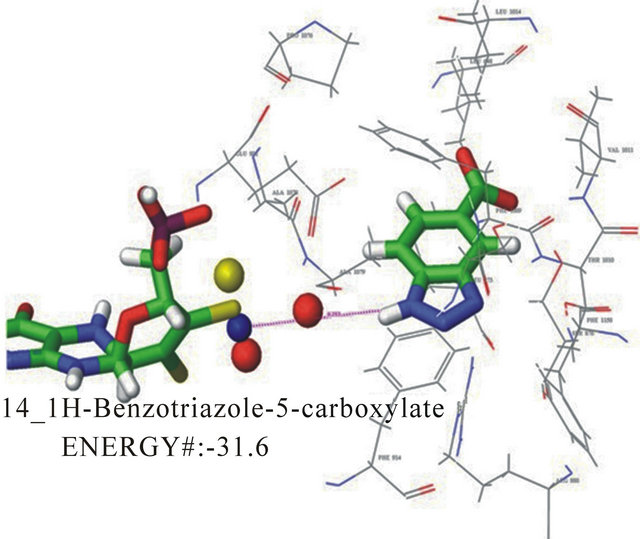
Figure 3. Docking pose A for 1H-benzotriazole-5-carboxylate with a docking energy of −31.6 kJ/mol
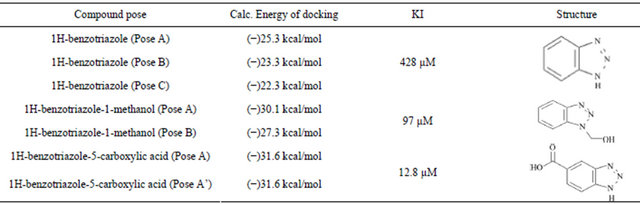
Table 1. The docking energies calculated for each pose of the compound correlated to the experimentally derived KI.
docking poses for this compound. The first pose has the hydroxyl group up and oriented towards the molybdenum cofactor. This pose also has electrostatic interacttions between the triazole nitrogen and arginine 880. Hydrogen bonding exists between the substituent and the molybdenum coordination sphere. This pose also has phenylalanine sandwiching with the inhibitor seen in Figure 4. The second pose (not shown) orients the carboxyl away from the molybdenum and down. The pose demonstrates phenylala nine sandwiching of the inhibitor as well. In the second pose the triazole nitrogen in oriented toward the molybdenum coordination sphere with the methanol group down, potentially interacting with arginine 880. The energies for these poses showed little difference with, −29.2 kJ/mol and −30.8 kJ/mol, respectively. This pose and kinetic data for 1H-benzotriazole-1- methanol suggests an extremely slow turnover and moderate overall catalytic inhibition. Based on the energy rankings, this compound uter docking estimations predicted 14 binding poses (not shown), for this compound suggesting little specificity and supports the high experimental value of KI, 428 mM. Interactions that do exist in each of these poses include the phenylalanine sandwiching of the inhibitor. This is as expected for a relatively small compound with few substituents.
All of the inhibitors maintained the hydrophobic interactions created by the phenylalanines. In general, triazole nitrogens were either oriented toward the molybdenum coordination sphere or towards arginine 880. the inhibitor ring
4. DISCUSSION
The results of the Glide® docking represented the docking energies of the various substrates within the active site. The docking energies represent electrostatic effects and hydrogen bonding, but do not include effects from entropy or hydrophobic interactions. For this reason the docking energies obtained from Glide® represent an overestimate of KI and are typically only used to rank
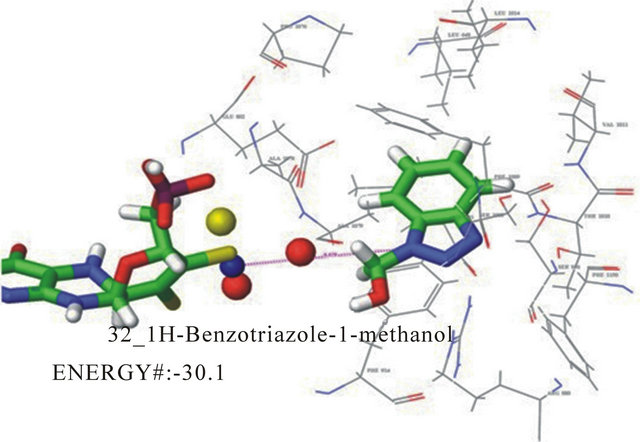
Figure 4. One of two docking poses for 1H-benzotriazole-1- methanol with a docking energy of −30.1 kJ/mol.
compounds for potential effectiveness. Energy ranking from docking results correlate well with steady state kinetic results obtained experimentally as shown in Table 1, thereby fulfilling the aims of the proposed hypothesis and revealing an effective rapid screening model for ranking potential effectiveness of a compound as an inhibitor or substrate. The docking results ranked 1Hbenzotriazole as the least specific compound in the active site which was evidenced by the 14 energetically favorable binding poses predicted. The expectation of this small unmodified 1H-benzotriazole was that it would bind in multiple orientations with little specificity due to the fact that no substituents were present to moderate binding orientation. 1H-benzotriazole-1-methanol was the second most specific inhibitor according to the docking model with two binding poses predicted with favorable docking energies. These two predicted binding poses represent drastically different orientations within the active site with equally favorable docking energies. 1Hbenzotriazole-5-carboxycylic acid ranked first in its docking energy which corresponded well with the smallest KI obtained kinetically. The docking program predicted five binding poses for this compound differing in the orientation of the carboxyl group. Both the neutral and charged forms of this compound were docked for comparison in this study. The docking orientation suggests that arginine 880 is involved in electrostatic interaction with the negatively charged carboxyl group. In the neutral form of the carboxyl group, the negative electron density from the triazole ring is stabilized by the positive charge on arginine 880.
Results from our study demonstrate the effectiveness of our model for correlating docking energies to experimental analyses. Our successful model is revealed at a time when the development of new inhibitors is still necessary and will serve as a method for testing these inhibitors rapidly without extensive kinetic experiments. Additionally, our model can be used to predict effectiveness of theoretical substrates of the enzyme. Exploration of substrate orientation is also possible, which was recently associated with the enzyme’s catalytic power [38] .
REFERENCES
- Choi, E.Y., Stockert, A.L., Leimkuhler, S. and Hille, R. (2004) Studies on the mechanism of action of xanthine oxidase. Journal of Inorganic Biochemistry, 98, 841-848. doi:10.1016/j.jinorgbio.2003.11.010
- Doonan, C.J., Stockert, A., Hille, R. and George, G.N. (2005) Nature of the catalytically labile oxygen at the active site of xanthine oxidase. Journal of the American Chemical Society, 127, 4518-4522. doi:10.1021/ja042500o
- Hemann, C., Ilich, P., Stockert, A.L., Choi, E.Y. and Hille, R. (2005) Resonance Raman studies of xanthine oxidase: The reduced enzyme-product complex with violapterin. The Journal of Physical Chemistry B, 109, 3023- 3031. doi:10.1021/jp046636k
- Hille, R. (1996) The mononuclear molybdenum enzymes. Chemical Reviews, 96, 2757-2816. doi:10.1021/cr950061t
- Hille, R. (1997) Mechanistic aspects of the mononuclear molybdenum enzymes. Journal of Biological Inorganic Chemistry, 2, 804-809. doi:10.1007/s007750050199
- Huber, R., Hof, P., Duarte, R.O., Moura, J.J.G., Moura, I., Liu, M.Y., LeGall, J., Hille, R., Archer, M. and Romao, M.J. (1996) A structure-based catalytic mechanism for the xanthine oxidase family of molybdenum enzymes. Proceedings of the National Academy of Sciences, 93, 8846- 8851. doi:10.1073/pnas.93.17.8846
- Kim, J.H., Ryan, M.G., Knaut, H. and Hille, R. (1996) The reductive half-reaction of xanthine oxidase. The involvement of prototropic equilibria in the course of the catalytic sequence. The Journal of Biological Chemistry, 271, 6771-6780.
- Okamoto, K., Matsumoto, K., Hille, R., Eger, B.T., Pai, E.F. and Nishino, T. (2004) The crystal structure of xanthine oxidoreductase during catalysis: Implications for reaction mechanism and enzyme inhibition. Proceedings of the National Academy of Sciences, 101, 7931-7936. doi:10.1073/pnas.0400973101
- Stockert, A.L., Shinde, S.S., Anderson, R.F. and Hille, R. (2002) The reaction mechanism of xanthine oxidase: Evidence for two-electron chemistry rather than sequential one-electron steps. Journal of the American Chemical Society, 124, 14554-14555. doi:10.1021/ja027388d
- Agarwal, A., Banerjee, A. and Banerjee, U.C. (2011) Xanthine oxidoreductase: A journey from purine metabo-lism to cardiovascular excitation-contraction coupling. Critical Reviews in Biotechnology, 31, 264-280. doi:10.3109/07388551.2010.527823
- Akalin, E., Ganeshan, S.V., Winston, J. and Muntner, P. (2008) Hyperuricemia is associated with the development of the composite outcomes of new cardiovascular events and chronic allograft nephropathy. Transplantation, 86, 652-658. doi:10.1097/TP.0b013e3181814f5b
- Angelos, M.G., Kutala, V.K., Torres, C.A., He, G.L., Stoner, J.D., Mohammad, M. and Kuppusamy, P. (2006) Hypoxic reperfusion of the ischemic heart and oxygen radical generation. American Journal of Physiology—Heart and Circulatory Physiology, 290, H341-H347. doi:10.1152/ajpheart.00223.2005
- Anker, S.D., Doehner, W., Rauchhaus, M., Sharma, R., Francis, D., Knosalla, C., Davos, C.H., Cicoira, M., Shamim, W., Kemp, M., Segal, R., Osterziel, K.J., Leyva, F., Hetzer, R., Ponikowski, P. and Coats, A.J.S. (2003) Uric acid and survival in chronic heart failure—Validation and application in metabolic, functional, and hemodynamic staging. Circulation, 107, 1991-1997. doi:10.1161/01.CIR.0000065637.10517.A0
- Becker, L.B., Vanden, H.T.L., Shao, Z.H., Li, C.Q. and Schumacker, P.T. (1999) Generation of superoxide in cardiomyocytes during ischemia before reperfusion. American Journal of Physiology—Heart and Circulatory Physiology, 277, H2240-H2246.
- Berry, C.E. and Hare, J.M. (2004) Xanthine oxicloreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. Journal of Physiology (London), 555, 589-606. doi:10.1113/jphysiol.2003.055913
- Braunersreuther, V. and Jaquet, V. (2012) Reactive oxygen species in myocardial reperfusion injury: From physiopathology to therapeutic approaches. Current Pharmaceutical Biotechnology, 13, 97-114. doi:10.2174/138920112798868782
- Cappola, T.P., Kass, D.A., Nelson, G.S., Berger, R.D., Rosas, G.O., Kobeissi, Z.A., Marban, E. and Hare, J.M. (2001) Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation, 104, 2407-2411. doi:10.1161/hc4501.098928
- Coghlan, J.G., Flitter, W.D., Clutton, S.M., Panda, R., Daly, R., Wright, G., Ilsley, C.D. and Slater, T.F. (1994) Allopurinol pretreatment improves postoperative recovery and reduces lipid-peroxidation in patients undergoing coronary-artery bypass-grafting. Journal of Thoracic and Cardiovascular Surgery, 107, 248-256.
- Anonymous (1966) Allopurinol for gout. British Medical Journal, 2, 317.
- Stockert, A.L. and Stechschulte, M. (2010) Allopurinol to febuxostat: How far have we come? Clinical Medicine Insights: Therapeutics, 2, 927-945.
- Fernandez, L., Heredia, N., Grande, L., Gomez, G., Rimola, A., Marco, A., Gelpi, E., Rosello-Catafau, J. and Peralta, C. (2002) Preconditioning protects liver and lung damage in rat liver transplantation: Role of xan-thine/ xanthine oxidase. Hepatology, 36, 562-572. doi:10.1053/jhep.2002.34616
- Grune, T., Schneider, W. and Siems, W.G. (1993) Reoxygenation injury of rat hepatocytes—Evaluation of nucleotide depletion and oxidative stress as causal components. Cell and Molecular Biology, 39, 635-650.
- Kang, S.M., Lim, S., Song, H., Chang, W., Lee, S., Bae, S., Chung, J. H., Lee, H., Kim, H.G., Yoon, D.H., Kim, T.W., Jang, Y., Sung, J.M., Chung, N.S. and Hwang, K.C. (2006) Allopurinol modulates reactive oxygen species generation and Ca2+ overload in ischemia-reperfused heart and hypoxia-reoxygenated cardiomyocytes. European Journal of Pharmacology, 535, 212-219. doi:10.1016/j.ejphar.2006.01.013
- Kinugasa, Y., Ogino, K., Furuse, Y., Shiomi, T., Tsutsui, H., Yamamoto, T., Igawa, O., Hisatome, I. and Shigemasa, C. (2003) Allopurinol improves cardiac dysfunction after ischemia-reperfusion via reduction of oxidative stress in isolated perfused rat hearts. Circulation Journal, 67, 781- 787. doi:10.1253/circj.67.781
- Lee, J., Hu, Q.S., Mansoor, A., Kamdar, F. and Zhang, J.Y. (2011) Effect of acute xanthine oxidase inhibition on myocardial energetics during basal and very high cardiac workstates. Journal of Cardiovascular Translational Research, 4, 504-513. doi:10.1007/s12265-011-9276-0
- Lee, W.Y. and Lee, S.M. (2006) Synergistic protective effect of ischemic preconditioning and allopurinol on ischemia/reperfusion injury in rat liver. Biochemical and Biophysical Research Communications, 349, 1087-1093. doi:10.1016/j.bbrc.2006.08.140
- Liu, P.G., He, S.Q., Zhang, Y.H. and Wu, J. (2008) Protective effects of apocynin and allopurinol on ische-mia/ reperfusion-induced liver injury in mice. World Journal of Gastroenterology, 14, 2832-2837. doi:10.3748/wjg.14.2832
- Matsumoto, F., Sakai, M., Yamaguchi, M., Nakano, H., Matsumiya, A., Kumada, K., Yoshida, K., Shimura, H., Machida, H., Takeuchi, S., Sasaya, S., Midorikawa, T. and Sanada, Y. (1997) Allopurinol reduced hepatic ischemia-reperfusion injury exacerbated by inhalation of high-concentration oxygen in rats. European Surgical Research, 29, 429-437. doi:10.1159/000129554
- Mubarak, H.A. (2011) Synergistic effect of ischemic preconditioning, postcoditioning and xanthine oxidase inhibition on cardiac tissue apoptosis of hepatic ischemic-reperfused male rats. Life Science Journal, 8, 253-262.
- Peglow, S., Toledo, A.H., Anaya-Prado, R., Lopez-Neblina, F. and Toledo-Pereyra, L.H. (2011) Al-lopurinol and xanthine oxidase inhibition in liver ischemia reperfusion. Journal of Hepato-Biliary-Pancreatic Sciences, 18, 137-146. doi:10.1007/s00534-010-0328-7
- Rhoden, E., Teloken, C., Lucas, M., Rhoden, C., Mauri, M., Zettler, C., Bello-Klein, A. and Barros, E. (2000) Protective effect of allopurinol in the renal ische-mia-reperfusion in uninephrectomized rats. General Pharmacology: The Vascular System, 35, 189-193. doi:10.1016/S0306-3623(01)00105-7
- Xiao, J., She, Q., Wang, Y., Luo, K.L., Yin, Y.H., Hu, R. and Huang, K.S. (2009) Effect of allopurinol on cardiomyocyte apoptosis in rats after myocardial infarction. European Journal of Heart Failure, 11, 20-27. doi:10.1093/eurjhf/hfn003
- (2005) Glide. Schroedinger, LLC.
- Zikakis, J.P. and Biasotto, N.O. (1976) Improved Isolation and purification method of xanthine-oxidase from bovine raw whole milk. Abstract of Papers American Chemical Society, 172, 140.
- Eger, B.T., Okamoto, K., Enroth, C., Sato, M., Nishino, T., Pai, E.F. and Nishino, T. (2000) Purification, crystallization and preliminary X-ray diffraction studies of xanthine dehydrogenase and xanthine oxidase isolated from bovine milk. Acta Crystallographica Section D, 56, 1656- 1658. doi:10.1107/S0907444900012890
- Okamoto, K., Eger, B.T., Nishino, T., Kondo, S., Pai, E.F. and Nishino, T. (2003) An extremely potent inhibitor of xanthine oxidoreductase—Crystal structure of the enzyme-inhibitor complex and mechanism of inhibition. The Journal of Biological Chemistry, 278, 1848-1855. doi:10.1074/jbc.M208307200
- Enroth, C., Eger, B.T., Okamoto, K., Nishino, T., Nishino, T. and Pai, E.F. (2000) Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: Structure-based mechanism of conversion. Proceedings of the National Academy of Sciences, 97, 10723-10728. doi:10.1073/pnas.97.20.10723
- Cao, H.N., Pauff, J. and Hille, R. (2011) Substrate orientation and the origin of catalytic power in xanthine oxidoreductase. Indian Journal of Chemistry A, 50, 355- 362.