Advances in Chemical Engineering and Science
Vol.2 No.1(2012), Article ID:16712,14 pages DOI:10.4236/aces.2012.21003
Low Molecular Weight Poly(Lactide-co-Caprolactone) for Tissue Adhesion and Tetracycline Hydrochloride Controlled Release in Wound Management
1Program in Petrochemistry and Polymer Science, Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok, Thailand
2Department of Chemistry, Faculty of Science, Chulalongkorn University, Bangkok, Thailand
3Institute of Biotechnology and Genetic Engineering, Chulalongkorn University, Bangkok, Thailand
Email: *Nuttha.T@chula.ac.th
Received September 17, 2011; revised October 25, 2011; accepted November 12, 2011
Keywords: L-Lactide; e-Caprolactone; Chitosan; Tissue Adhesive; Tetracycline Hydrochloride Release; Co-Initiator; Wound Healing
ABSTRACT
The use of biopolymers as bioadhesives for human tissue is becoming a preferred alternative to suturing due to their superior adhesive, biocompatible, and biodegradable properties. In this work, low molecular weight poly(L-lactide-coe-caprolactone) (P(LA-co-CL) was synthesized to achieve the glass transition temperature (Tg) of the copolymer at ambient temperature so that during application on the skin, the copolymer when combined with chitosan (CHI) into the CHI/P(LA-co-CL) film could provide the strong support at the injury site. Using alcohols with different numbers of hydroxyl groups as the co-initiator in polymerization provided the distinctive characteristics of copolymers. Among all copolymers synthesized, P(LA-co-CL) copolymer using pentaerythritol as the co-initiator when combined with CHI at the ratio of copolymer/CHI at 70/30 yielded the good film properties in tissue adhesion and tetracycline hydrochloride release.
1. Introduction
The skin is often known as the largest organ of the human body. The skin is composed of 2 major layers of tissue. The outer epidermis provides protection and prevents foreign materials including microorganisms from entering the body. The inner dermis consists of 3 main components including 1) blood vessels, 2) accessory structures (e.g. hair, nails, sweat glands, and sebaceous glands), and 3) collagen and elastin which give the skin its strength and flexibility. In normal skin, the epidermis and dermis form a protective barrier against the external environment at steady state equilibrium. Once the protective barrier is broken, the physiological process of wound healing is immediately resumed. Wound repair or wound healing is a complicated process in which the skin repairs itself after injury. The classical model of wound healing is divided into 4 phases, i.e., hemostasis, inflammatory, proliferative, and remodeling.
Upon injury to the skin, complex biochemical reactions occur systematically to repair the damage. Within minutes after injury, platelets (thrombocytes) aggregate at the injury site to form a fibrin clot. This clot controls active bleeding (hemostasis). In the inflammatory phase, bacteria and debris at the injury site are phagocytosed and removed, and factors are released that lead to migration and division of cells involved in the proliferative phase. In the proliferative phase, new blood vessels are formed by vascular endothelial cells; fibroblasts grow and form a new extracellular matrix (ECM) by excreting collagen and fibronectin. At the same time, re-epithelialization of the epidermis occurs, providing cover for the new tissue. In contraction, myofibroblasts establish a grip on the wound edges and contract themselves using a mechanism similar to that in smooth muscle cells; thus making the wound become smaller. When the cells’ roles are readily completed, unneeded cells undergo apoptosis. In maturation and remodeling phase, collagen is remodeled and realigned along tension lines and cells that are no longer needed are removed by apoptosis. Nevertheless, wound healing process is not only complex but delicate, and susceptible to interruption or failure leading to a chronic non-healing wound. Factors which may contribute to this failure include diabetes, venous or arterial disease, aging, and infection.
To prevent the failure during wound healing process, wound management aids are important to promote the healing process. Wound management is performed by initial cleansing and debridement. After antiseptic procedures are completed, the wound has to be covered with dressing [1]. An ideal wound dressing should provide the optimal environment to meet the treatment objective and protection from further injury. Table 1 shows the characteristics of the ideal wound dressing [2,3]. Generally, the dressing should be capable of speeding up the healing process, preventing infection, and avoiding second injury at the end of healing process. In addition, an antibiotic is commonly applied to the dressing for reducing inflammation caused by bacterial infection. There are several dressing products commercially available which possess different description and indication. The examples of the adhesives those are currently in clinical use include fibrin sealants, albumin-based compounds, cyanoacrylates, hydrogels, and collagen-based adhesives [4]. Although those dressing devices exhibited the satisfactory performance, the application of an inappropriate dressing may adversely affect wound healing process. Therefore, care must be taken during wound management.
Currently, the use of biopolymers as bioadhesives for human tissue is emerging as a preferred alternative to suturing [5-9]. Those biopolymers serve as the ideal dressing since they are capable of providing sufficient viscosity; thus adhere to the tissue substrates or wound rapidly. They are also biocompatible; thus, do not lead to the systematic toxicity [10,11]. However, those polymers including chitosan (CHI), polylactic acid (PLA), and polycaprolactone (PCL) could not serve all ideal dressing characteristics mentioned above when each was fabriccated into the dressing device alone. Although CHI is mentioned to be the distinctively wound healing facilitator, it usually suffers from its swelling property especially when applying on the bleeding wound. PLA provides the good mechanical strength but lacks heat resistant property. PCL is biocompatible but its thermal properties are rather poor. With the good ratio of each polymer in the composite, it was expected that the dressing prepared would protect wound from dryness and second injury during daily cleansing besides promoting the self treatment process by body. Therefore, in this work, low molecular weight poly(L-lactide-co-e-caprolactone) was synthesized by ring opening polymerization to achieve the glass transition temperature (Tg) of the copolymer at approximately 20˚C - 25˚C. At this expected Tg, the change in viscosity of the copolymer was expected; thus, during application on the skin the copolymer fluidity could hold the injury site and provide the strong support for fibrin formation to facilitate self healing process. Furthermore, the addition of CHI and tetracycline hydrochloride helped prevent skin inflammatory from bacterial infection.
2. Materials and Methods
2.1. Synthesis of L-Lactate Oligomer
L-lactic acid solution was heated at 140˚C for 2 h under a dry nitrogen atmosphere. High vacuum was applied during this step for 30 min to eliminate the water initially presented in the solution and that obtained by polymerization. After that the temperature was increased to 160˚C and the reaction mixture was maintained at this temperature for 3 h to obtain the resulting low molecular weight poly(L-lactic acid) (PLLA) or L-lactate oligomer.
2.2. L-Lactide Ring Formation
To form L-lactide, L-lactate oligomer was heated with zinc (at the weight ratio of 100:1) at 160˚C for 30 min. After that high vacuum was applied and the reaction mixture was heated to 220˚C - 230˚C until L-lactide crystal was formed. L-lactide crystal was removed and washed with cold deionized water to remove excess Llactic acid and then vacuum filtered. L-lactide crude was purified by dissolved in warm ethyl acetate to remove

Table 1. Characteristics of the ideal wound dressing [2].
insoluble materials and filtered. The filtrate was then heated to evaporate the remaining ethyl acetate and precipitate white needle-like crystal of L-lactide. L-lactide crystal was further purified twice according to the procedure mentioned above and dried in vacuum oven for 24 h prior to use.
2.3. Synthesis of Low Molecular Weight Poly(Lactide-co-Caprolactone)
Low molecular weight copolymers of L-lactide (LA) and ε-caprolactone (CL) were prepared using alcohols with different numbers of hydroxyl groups (1,4-butanediol (BD), pentaerythritol (PTOL), and D-sorbitol (SB)) as the co-initiator by stepwise ring opening polymerization. L-lactide and co-initiator was added into the three-neck round bottomed flask containing a magnetic bar and sealed with a rubber septum. The reaction was carried out at 145˚C for 150 min per block under the dry nitrogen atmosphere. Stannous(II) 2-ethylhexanoate (Sn(Oct)2) in toluene was added at a 1/400 catalyst/lactone molar ratio when the monomer began to melt. After that ε-caprolactone was added into the reaction and the reaction was carried out using the same procedure mentioned above. The reaction was completed when the appropriate arm length of copolymer was achieved. The crude products were dissolved in chloroform, precipitated several times in methanol, and dried in vacuum oven at 30˚C for 24 h. The resulting copolymer was kept under vacuum at ambient temperature until use.
2.4. Copolymer Characterization
The resulting copolymers were characterized by proton nuclear magnetic resonance spectrometer, gel permeation chromatography, and differential scanning calorimeter.
2.5. Proton Nuclear Magnetic Resonance Spectrometer (1H NMR)
Proton (1H) nuclear magnetic resonance analysis was used to characterize L-lactide and copolymer products. The sample was dissolved in chloroform-d (CDCl3) and vortexed until clear solution was obtained. The NMR experiment was carried out using Varian mercury-400 spectrometer 1H NMR operating at 400 MHz. Chemical shifts (d) were reported in parts per million (ppm) relative to the residual protonated solvent signal as a reference.
2.6. Gel Permeation Chromatograph (GPC)
Gel permeation chromatography (GPC) was used to determine the molecular weight of copolymer products ( and
and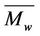 ) and polydispersity index (PDI). The copolymer sample was dissolved in tetrahydrofura (THF) and filtered by the syringe nylon filter (0.45 µm). GPC chromatogram of copolymer was obtained from Waters 150-CV chromatography equipped with PL-gel 10 µm mixed B 2 columns (MW resolving range of 500 - 10,000,000) at 35˚C. THF was used as an eluent at the flow rate of 1.0 mL/min. Before injecting the sample, degassed THF was passed through the column for 20 min. 100 µL sample was injected and run for 40 min. The molecular weight was determined by a reflection index detector using Polystyrenes (MW of 5460 - 1,290,000) as the standards for calibration.
) and polydispersity index (PDI). The copolymer sample was dissolved in tetrahydrofura (THF) and filtered by the syringe nylon filter (0.45 µm). GPC chromatogram of copolymer was obtained from Waters 150-CV chromatography equipped with PL-gel 10 µm mixed B 2 columns (MW resolving range of 500 - 10,000,000) at 35˚C. THF was used as an eluent at the flow rate of 1.0 mL/min. Before injecting the sample, degassed THF was passed through the column for 20 min. 100 µL sample was injected and run for 40 min. The molecular weight was determined by a reflection index detector using Polystyrenes (MW of 5460 - 1,290,000) as the standards for calibration.
2.7. Differential Scanning Calorimeter (DSC)
DSC (NETZSCH DSC204F1 Phoenix) was used to examine the glass transition as a change in the heat capacity as the polymer matrix went from the glass state to the rubber state. Copolymer was weighed in a sample pan. The starting temperature was at 20˚C. It was heated to 160˚C with the heating rate of 20˚C/min and remained isothermal for 3 min. In this step, copolymer would be completely molten. Liquid nitrogen was used to reduce temperature until –50˚C with the cooling rate of 20˚C/ min and kept isothermal for 3 min. The semi-crystalline structure was precipitated rapidly while the amorphous region could not be precipitated within a short time. The sample was heated to 200˚C (heating rate 20˚C/min) until completely molten again. The empty pan was used as standard calibration. Tg was defined as the temperature that the amorphous region could be changed from glasslike to rubber-like.
2.8. Preparation of Blended CHI/P(LA-co-CL) Films
CHI flakes were dissolved into 1% (v/v) acetic acid to obtain the homogeneous CHI solution (2% CHI). P(LAco-CL) particles were dissolved into a mixture of acetone and ethanol (acetone/ethanol at 4/1 (v/v)). CHI/P(LA-coCL) blended film was prepared by the modified spontaneous emulsification-solvent diffusion method (modified SE-SD method) [12]. P(LA-co-CL) solution was slowly dropped onto the CHI solution (2%) containing 0.5% (w/w) glycerol while the reaction mixture was continuously stirred at 600 rpm to obtain the P(LA-co-CL) particle suspension into CHI solution. After that the reaction mixture was pour onto the Petri dish to allow film casting after solvent evaporation. The resulting film was dried at 30˚C for 72 h and kept under vacuum at ambient temperature for further characterization and use.
2.9. Morphology of CHI/P(LA-co-CL) Blended Films
The thickness of the CHI/P(LA-co-CL) blended film was measured by a micrometer at 5 different locations (at the center and the corners). The thickness mean was calculated from 3 samples.
Film surface morphology was observed under the scanning electron microscope (SEM) (Jeol JSM-5800 LV). The prepared films were mounted on the metal grids with double-sided adhesive tape and coated with gold under high vacuum before observation.
2.10. Swelling Property
The swelling property of the films was determined by measuring the water adsorption. The films were cut into 2 × 2 cm2 and weighed before immerged into the glass bottles containing 50 mL phosphate buffer saline (PBS) (pH 7.4) at ambient temperature. At an appropriate time interval, the films were taken out, and the excess water was removed carefully with filter paper. Then the films were weighed immediately. The degree of swelling was determined in Equation (1):
 (1)
(1)
where Wt is the weight of film at time t, and W0 is the weight of film at time zero.
2.11. In Vitro Tissue Adhesion
T-peel test was used to determine the adhesion property of the prepared films. The adhesive strength test was in vitro performed using 30 µm thick polyamide (6/6 Nylon) films as substrates. The film sample (2.5 × 5 cm2) was adhered to substrate and the tested specimen (substrate with adhered film) was cut into 2.5 × 10 cm2. T-Peel Test (ASTM D 1876) was performed using an Instron machine (model 5583). The measurement was performed at a 30 mm/min crosshead speed at 37˚C. Each test was repeated 3 times. Adhesive Failure Strength (AFS) values were determined and reported in Newton per centimeter width units (N/cm width).
2.12. In Vitro Tetracycline Hydrochloride (TCH) Release Study
TCH was loaded onto the CHI/P(LA-co-CL) blended films at the ratios of 2.5%, 5%, and 10% (w/w) , respectively. The TCH loaded blended film was cut into the square shape (30 mm × 30 mm × 0.1 mm, with approx. 80 mg weight). The film was immersed into the flask containing 50 mL PBS (pH 7.4). The flask was then incubated at 37˚C and 110 rpm. 3 mL sample was taken from the flask and 3 mL fresh PBS was replaced to maintain the constant volume during the test. The sample was centrifuged and diluted before measuring UV absorption at 276.7 nm (UV-Vis spectrophotometer). The amount of TCH released was calculated by interpolation from a calibration curve (TCH concentration versus UV absorption at 276.7 nm). A cumulative correction was also made for the previously removed sample to determine the total amount of drug release. All experiments were performed 3 times.
3. Results and Discussion
3.1. Synthesis and Characterization of L-Lactide and P(LA-co-CL)
Figure 1 shows the characteristic of L-lactide crystal by 1H NMR technique. From 1H NMR spectrum, the chemical shift at 5.07 ppm was assigned for the methine proton (-CH-CH3), and 1.66 ppm for methyl protons (-CHCH3).
The series of low molecular weight (LMW) copolymers of LA and CL possessing the variations in arm number and arm length were synthesized through stepwise ring opening polymerization of LA and CL using alcohols with different numbers of hydroxyl groups in the presence of Sn(Oct)2 under nitrogen atmosphere. After the reaction was completed, the crude copolymer turned into yellowish viscous liquid and then it was allowed to solidify at room temperature. The yield percentages and the physical appearances of purified LMW copolymers are shown in Table 2. It was found that the structure of the copolymers obtained was dependent on the functionality of the alcohol used as the co-initiator. Bifunctional alcohols yielded linear copolymers (BD) while alcohols with the OH groups higher than 2 gave branched and star-shaped P(LA-co-CL) copolymers (PTOL and SB) (Figure 2).
By controlling the monomer to co-initiator molar ratio, the differences in arm length and molecular weight were

Figure 1. 1H NMR spectrum of L-lactide.

Table 2. Yields and physical appearances of low molecular weight copolymers of L-lactide and e-caprolactone synthesized by stepwise ring opening polymerization with the presence of alcohols at different numbers of hydroxyl groups.

Figure 2. Structures of low molecular weight copolymers with similar arm length (a) and similar molecular weight (b). (a) Similar arm length; (b) Similar molecular weight (~10,000 g/mol).
achieved. Copolymers with similar arm length contained 24 LA units and 4 CL units. On the other hand, copolymers with similar molecular weight (10,000 g/mol), arm length decreased when the OH groups of co-initiator increased. The resulting copolymers were characterized by 1H NMR, GPC and DSC.
3.2. Proton Nuclear Magnetic Resonance Spectroscopy (1H NMR) Analysis
An example of 1H NMR spectrum of two-arm or linear P(LA-co-CL) is shown in Figure 3. The signal of the methine proton (-CH-CH3, a) of PLLA repeat unit is at 5.20 ppm. The methine proton (-CH-OH, b) of the chain end unit is at 4.35 ppm. The methylene protons of PCL repeat unit are at 4.00 - 4.20 (-CH2-O-, c) and 2.23 - 2.41 ppm (-CH2-COO-, d), respectively. The methyl group (-CH-CH3, e) of PLLA repeat unit and the methylene protons (-CH2-CH2-, f and g) of PCL repeat unit are at 1.37 - 1.70 ppm. Similarly, copolymerization of LA and CL with PTOL and SB provided well-defined four, sixarm, star-shaped P(LA-co-CL). The four and six-arm, star-shaped copolymers showed the typical proton signals of the P(LA-co-CL) main chain as observed in twoarm copolymer.
From 1H NMR results, monomer ratio, arm length, number of hydroxyl group, and molecular weight could be calculated from the integral peaks [13]. From the peak area integrations, the copolymer compositions can be determined as showed in Table 3. It was found that the actual LA to CL ratios of the copolymer obtained was lower than the calculated theoretical value because the trace amounts of moisture affected the molecular weight and composition of the copolymer. Linear P(LA-co-CL) (CO-BD) had a theoretical chain length of 24 LA units and 4 CL units whereas the chain length determined by 1H NMR was 23.5 and 5.75, respectively. The results were corresponded to the value of 1.91 as the average number of OH groups initiating polymerization. This was also similar to four-arm P(LA-co-CL) (CO-PTOL) (Table 3). However, the actual chain length of the six-arm P(LA-co-CL) (CO-SB) was larger than the theoretical value. This was probably due to the lower activity of the secondary OH groups [13].
3.3. Gel Permeation Chromatography (GPC) Analysis
The ,
,  , and PDI of P(LA-co-CL) copolymers are shown in Table 4. It was observed that increasing the
, and PDI of P(LA-co-CL) copolymers are shown in Table 4. It was observed that increasing the

Figure 3. 1H NMR spectrum of P(LA-co-CL) using 1,4-butanediol as co-initiator (CO-BD).

Table 3. Compositions and chemical characteristics of low molecular weight copolymers of L-lactide and e-caprolactone synthesized by stepwise ring opening polymerization with the presence of alcohols at different numbers of hydroxyl groups.

Table 4. Properties of low molecular weight copolymers of L-lactide and e-caprolactone synthesized by stepwise ring opening polymerization with the presence of alcohols at different numbers of hydroxyl groups.
number of OH groups in the copolymer with similar chain length increased the molecular weight of copolymer. For all copolymers, the  determined by GPC was closely corresponded to the theoretical values. In addition, excellent agreement existed between the 1H NMR and GPC techniques. The
determined by GPC was closely corresponded to the theoretical values. In addition, excellent agreement existed between the 1H NMR and GPC techniques. The  was in agreement with the targeted molecular weight. The PDIs for the linear, four-arm, and star-shaped P(LA-co-CL) copolymers were narrow while the PDIs for the six-arm and star-shaped P(LA-co-CL) copolymers were relatively broad. It was possibly due to the increasing stearic hindrance with increasing arm molar mass, number of arms, and transesterification reactions. From 1H NMR and GPC, this copolymer composition is similar to the LA and CL feed mole ratio. Therefore, the synthesized reaction was successfully taken to near-quantitative conversion.
was in agreement with the targeted molecular weight. The PDIs for the linear, four-arm, and star-shaped P(LA-co-CL) copolymers were narrow while the PDIs for the six-arm and star-shaped P(LA-co-CL) copolymers were relatively broad. It was possibly due to the increasing stearic hindrance with increasing arm molar mass, number of arms, and transesterification reactions. From 1H NMR and GPC, this copolymer composition is similar to the LA and CL feed mole ratio. Therefore, the synthesized reaction was successfully taken to near-quantitative conversion.
3.4. Differential Scanning Calorimetry (DSC) Analysis
It was expected that the Tg of the synthesized copolymers could be between 20˚C - 25˚C so that the copolymers became more fluidity at the temperature above those values (such as at 37˚C—the body temperature). As the result, the copolymer could hold the injured tissue. According to the thermal properties of homopolymer, PLLA contains about 37% crystalline region corresponded to the melting point of 175˚C - 178˚C and the Tg of 60˚C - 65˚C. Whereas PCL is a semicrystalline polymer with the melting point of 59˚C - 64˚C and the Tg of –60˚C [14]. Therefore, the copolymer Tg was decreased with the incorporation of caprolactone molecules into the chains of lactide. The Tg of copolymers are shown in Table 4. It was found that the Tg of copolymers change with the copolymers composition. With increase of LA/CL ratio, the Tg of copolymer increased from 10.8˚C to 31.6˚C. The Tgs of the CO-BD, CO-PTOL, and CO-PTOL copolymers with the similar arm length (24/4, LA/CL), using BD, PTOL and SB were at 21.4˚C, 22.8˚C, and 25.9˚C, respectively. On the other hand, the Tgs of the CO-BD (10,000) and CO-SB (10,000) copolymers with the similar molecular weight (~10,000 g/mol) were at 31.6˚C and 10.8˚C, respectively. It was found that the Tg of CO-BD (10,000) was higher than the expected temperature due to the high LA/CL ratio while the Tg of CO-SB (10,000) was lower than the expected temperature because of the low LA/CL ratio. These results confirmed that the Tg of copolymer was increased with the increasing LA/CL ratio.
3.5. CHI/P(LA-co-CL) Blended Films and Their Properties
CHI/P(LA-co-CL) films were prepared by the modified-SESD method at the different polymer compositions (Table 5). The appearances of the sample films are shown in Table 5 and Figure 4. The most appropriate copolymer to CHI ratio in preparing the CO-BD/CHI blended film was at 70/30. It is obvious that the presence of CHI in the film helped improve the flexibility of the films due to the high flexibility characteristic of CHI.
The thickness of the blended films obtained was measured and found to be in the range of 0.10 - 0.15 mm. The film surface was investigated under the SEM as seen in Figure 5. Similar morphology was observed in all blended films. It was observed that the surfaces of the blended films (Figure 5) were rougher and more opaque than that of the CHI film (data not shown). The pore sizes observed on the film surfaces were approximately 0.5 - 1.0 µm.
TCH loaded CHI/P(LA-co-CL) blended film was also prepared. During the preparation TCH was partially dissolved into CHI matrix and incorporated in the copolymer pore. TCH seemed to exhibit no effect on film surface morphology as observed in Figure 6(a). However, when comparing the cross sectional structures of the blended films with and without the presence of TCH (Figures 6(b) and (c)), it was found that the incorporation of TCH gave the higher intra porosity of the blended film. This was perhaps because the ionic interaction between the  groups of CHI and the COO– groups of
groups of CHI and the COO– groups of

Figure 4. Appearances of CHI/P(LA-co-CL) blended films at various copolymer to CHI ratios, i.e., CO-BD/CHI at 70/ 30 (0.5 g) (a); CO-BD/CHI at 70/30 (1.0 g) (b); CO-BD/CHI at 80/20 (0.5 g) (c); CO-BD/CHI at 80/20 (1.0 g) (d); COBD/CHI at 90/10 (0.5 g) (e); and CO-BD/CHI at 90/10 (1.0 g) (f).

Table 5. Formation of CHI/P(LA-co-CL) blended films at various copolymer to CHI ratios.
TCH formed the strong network when the drug was incorporated in the blended film. The shelf life of the prepared film was investigated. It was found that the film morphology changed after storage for 1 month (data not shown). The short shelf life of the blended film prepared led to the limited use in commercialization; thus, it is necessary to improve this storage property. Besides, certain storage condition requires being determined later.

Figure 5. Scanning electron micrographs of the CHI/P(LAco-CL) blended films at the copolymer to CHI ratio of 70/30; the blended films containing the copolymer with the similar arm length (a)-(c); and the blended films containing the copolymer with the similar molecular weight (d)-(f) (at magnification of ×1000 at the left side and ×10,000 at the right side).

Figure 6. Scanning electron micrographs of the CHI/P(LAco-CL) blended films at the copolymer to CHI ratio of 70/30; the surface of TCH loaded CO-PTOL/CHI film at magnification of ×1000 at the left side and ×10,000 at the right side (a); cross section of TCH loaded CO-PTOL/CHI film (b); and cross section of CO-PTOL/CHI film (c).
The swelling behavior of various CHI/P(LA-co-CL) blended films in PBS buffer (pH 7.4) is demonstrated in Figure 7. It was found that CHI film exhibited the highest degree of swelling followed by the CO-PTOL/CHI (30/70) film. While the swelling degree of the copolymer/CHI (70/30) blended films at similar arm length or molecular weight are relatively similar (approximately 200% - 300%) and they were lower than those observed in CHI and CO-PTOL/CHI (30/70) films.
The blended films at the higher copolymer content lowered the swelling capacity of CHI present due to the stronger hydrophobicity in the copolymer matrices. This eventually led to less water penetration into the film structure. While the CHI films without the copolymer exhibited the higher swelling capacity. This could be explained by the flexibility of the CHI chains created more space for water within the polymer matrix. As a result, CHI structure became mechanically fragile. The results found in this study are correspondent to the claim that the swelling behavior was mainly dependent on the film structure and composition [15].
3.6. In Vitro Tissue Adhesion
It was claimed that the adhesive property of the blended film was dependent on the LA component and the chain entanglement [10]. As a consequence, the copolymers with the differences in arm length and molecular weight might exhibit distinct adhesive strength. In this study, all blended films were tested for adhesive failure strength (AFS) as seen in Table 6. The blended film with CO-BD gave the maximum AFS value while the film with COBD (10,000) yielded the lowest AFS value. Comparing

Figure 7. Swelling behavior of CHI/P(LA-co-CL) blended films in PBS buffer (pH 7.4).

Table 6. Adhesive failure strength (AFS) of the CHI/P(LAco-CL) blended films containing the copolymer/CHI ratio of 70/30.
the blended films containing the copolymers with the similar arm length (CO-BD/CHI (linear), CO-PTOL/CHI (branch), and CO-SB/CHI (star-shape)), increasing the arm numbers led to an increase in AFS value. The CO-PTOL/CHI and CO-SB/CHI blended films gave the similar AFS values. This was perhaps due to the actual numbers of the initiating OH (4.67 in Table 3) in the CO-SB copolymer.
Among the blended films with the similar molecular weight (different arm length), the highest AFS value was found in the film containing the linear copolymer (COBD (10,000)/CHI) perhaps due to the high Tg of the copolymer present in the blended film (Table 4). The AFS result was in agreement with the Tg result that confirmed no change in viscosity at the body temperature when applying the film at the injury site. In addition, decreasing arm length (or increasing the arm numbers) led to the increasing AFS values in the blended films containing the copolymers with the similar molecular weight.
Comparing the blended films containing the copolymers with the similar shape but different arm length (for example, CO-PTOL/CHI and CO-PTOL (10,000)/CHI), it was found that the film containing the copolymer with the longer arm length yielded the higher AFS value.
Among all films tested, the (CO-PTOL/CHI) film provided the highest AFS value; therefore, it would be prepared for the in vitro TCH release.
3.7. In Vitro Tetracycline Hydrochloride Release Study
TCH was used as a model drug to investigate the ability of the blended film prepared for controlled delivery of the desired active ingredient. Different TCH loading percentages were designed to modulate the optimal desorption rate in order to determine the small loading dosage and minimize numbers of administration during wound healing process (Table 7) [16]. The TCH dissolution profiles of the CHI/P(LA-co-CL) blended films are shown in Figure 8. Similar TCH release profile was observed from the CO-PTOL/CHI (70/30) blended film containing different TCH loads (Table 7 and Figure 8). TCH was rapidly released during the initial period (0 - 0.25 h). After that the release rate dropped. Increasing the loaded TCH percentage yielded more TCH adhered onto the film surface as well as that embedded in the film matrix [17]. TCH adhered at the film surface was predominantly released during the initial period. This claim was consistent with the findings in this study that a higher release rate during the initial period was observed at a higher TCH loading (Table 7). TCH release percentages at equilibrium observed from the CO-PTOL/ CHI (70/30) blended films containing different TCH loading were not different (Figure 8). This was probably because the degree of swelling of the CO-PTOL/CHI (70/30) blended film controlled the release percentage [17].
Among 3 films loaded with 5.0% TCH observed from Figure 8, it is clear that CHI film gave the highest TCH release percentage (98.28%) at equilibrium followed by CO-PTOL/CHI film (84.08%). Whereas CO-PTOL (10,000)/CHI yielded the lowest release rate (74.13%). This result was consistent with the film swelling capacity (Figure 7). CHI film possessed the largest degree of swelling (718.28%) while subsequently lower degree of swelling was observed in CO-PTOL/CHI (309.35%) and CO-PTOL (10,000)/CHI (202.17%) films, respectively. At higher degree of swelling, more TCH particles adhered on the film surface or embedded in the film structure are susceptible to dissolution; thus, leading to the higher release rate. In addition, the longer arm length of CO-PTOL present in the film might be responsible on the higher release rate compared with that observed in COPTOL (10,000)/CHI film.
From the results shown above, it is clear that the copolymer present in the blended film help lower TCH release rate. TCH release profiles observed from all films tested were also compared with a commercially available drug delivery system (ActisiteÒ, Alza Corporation, Palo Alto, CA), poly(ethylene-co-vinyl acetate) containing 25.0% TCH loading (Table 8). Initially, the release rates of both the CO-PTOL/CHI blended film and Actisite® were high. However, at equilibrium approximately 83.55% TCH was released from the CO-PTOL/CHI film while only 30% TCH was liberated from Actisite®.
4. Conclusions
In this work, P(LA-co-CL) copolymers with similar arm length and similar molecular weight were successfully synthesized via stepwise ring opening polymerization with the help of co-initiator (BD, PTOL, and SB). The 1H NMR results show that all synthesized copolymers contained nearly the same LA/CL mole fractions as those theoretical values calculated except when SB was used as the co-initiator. The ,
,  , and PDI of the copolymers were closed to the predicted values. The PDI of the linear and four-arm copolymers was narrow while relatively broad PDI was observed in the six-arm, starshaped copolymers. Copolymers CO-BD, CO-PTOL, COSB, and CO-PTOL (10,000) yielded the preferable Tg values of 21.4˚C, 22.8˚C, 25.9˚C, and 20.6˚C, respectively since at ambient or normal body temperature, the films containing such above mentioned copolymer exhibited more fluidity behavior; thus, they could hold the injured skin and facilitate wound healing process.
, and PDI of the copolymers were closed to the predicted values. The PDI of the linear and four-arm copolymers was narrow while relatively broad PDI was observed in the six-arm, starshaped copolymers. Copolymers CO-BD, CO-PTOL, COSB, and CO-PTOL (10,000) yielded the preferable Tg values of 21.4˚C, 22.8˚C, 25.9˚C, and 20.6˚C, respectively since at ambient or normal body temperature, the films containing such above mentioned copolymer exhibited more fluidity behavior; thus, they could hold the injured skin and facilitate wound healing process.
During film preparation, copolymer was incorporated

Figure 8. Dissolution profiles of TCH in PBS buffer (pH 7.4) at 37˚C from CHI/P(LA-co-CL) blended films containing various TCH loading.

Table 7. TCH release rates from the CHI/P(LA-co-CL) blended films containing CO-PTOL/CHI of 70/30 in PBS buffer (pH 7.4) at 37˚C.

Table 8. Specification of the TCH loaded CHI/P(LA-co-CL) film containing CO-PTOL/CHI at the ratio of 70/30 compared with the commercially available drug release device Actisite®.
into CHI with the suitable ratio of copolymer/CHI at 70/30 by weight. From the SEM micrographs, copolymer particles were dispersed into CHI matrix. Also, unexpected pores were observed at the surface of the blended films. Adhesive strength of blend films was investigated by T-peel test. According to the most appropriate Tg and AFS values, CO-PTOL/CHI and CO-PTOL (10,000)/ CHI films were selected for the in vitro TCH release study. In general, the mechanism of TCH release from copolymer/CHI film could be explained by TCH diffusion when film was swelling. At different amounts of TCH loading (2.5%, 5.0%, and 10.0%), the similar TCH dissolution profiles from the CO-PTOL/CHI films were observed. Comparing the dissolution profiles from the same TCH loading (5.0%) into CO-PTOL/CHI and COPTOL (10,000)/CHI films, it was found that CO-PTOL/ CHI film gave more proper release rates during the whole period observed. According to the results mentioned above, the CHI/P(LA-co-CL) film with the COPTOL/CHI ratio of 70/30 was best suited for further development into the wound dressing device.
5. Acknowledgements
This work has been partially supported by National Research University Project of Commission on Higher Education (CHE) and the Ratchadapiseksomphot Endowment Fund (AM1026A).
REFERENCES
- E. R. Kenawy, G. L. Bowlin, K. Mansfield, J. Layman, D. G. Simpsonc, E. H. Sandersb and G. E. Wneka, “Release of Tetracycline Hydrochloride from Electrospun Poly(Ethylene-co-Vinylacetate), Poly(Lactic Acid), and a Blend,” Journal of Controlled Release, Vol. 81, No. 1-2, 2002, pp. 57-64. doi:10.1016/S0168-3659(02)00041-X
- Northern Health and Social Services Board, “NHSSB Wound Management Manual,” Database on the Internet, 2011. http://www.nhssb.n-i.nhs.uk/publications/primary_care/Wound_Manual.pdf
- D. T. Foster and L. J. Rowedder, “Reese SK. Management of Sports-Induced Skin Wounds,” Journal of Athletic Training, Vol. 30, No. 2, 1995, pp. 135-144.
- L. Montanaro, C. R. Arciola, E. Cenni, G. Ciapetti, F. Savioli, F. Filippini and L. A. Barasanti, “Cytotoxicity, Blood Compatibility and Antimicrobial Activity of Two Cyanoacrylate Glues for Surgical Use,” Biomaterials, Vol. 22, No. 1, 2001, pp. 59-66. doi:10.1016/S0142-9612(00)00163-0
- S. K. Agrawal, N. S. DeLong, J. M. Coburn, G. N. Tew and S. R. Bhatia, “Novel Drug Release Profiles from Micellar Solutions of PLA-PEO-PLA Triblock Copolymers,” Journal of Controlled Release, Vol. 112, No. 1, 2006, pp. 64-71. doi:10.1016/j.jconrel.2005.12.024
- A. Gallardo, J. L. Eguiburu, M. J. Fernandez Berridi and J. S. Roman, “Preparation and in Vitro Release Studies of Ibuprofen-Loaded Films and Microspheres Made from Graft Copolymers of Poly(L-Lactic Acid) on Acrylic Backbones,” Journal of Controlled Release, Vol. 55, No. 2-3, 1998, pp. 171-179. doi:10.1016/S0168-3659(98)00047-9
- J. H. Lee, A. K. Go, S. H. Oh, K. E. Lee and S. H. Yuk, “Tissue Anti-Adhesion Potential of Ibuprofen-Loaded PLLA-PEG Diblock Copolymer Films,” Biomaterials, Vol. 26, No. 6, 2005, pp. 671-678. doi:10.1016/j.biomaterials.2004.03.009
- L. B. Peppas, “Polymers in Controlled Drug Delivery,” Biomaterials, 1997, p. 42. http://www.scribd.com/doc/15042915/polymer-in-drug-delivery.
- F. Wang, T. Lee and C. H. Wang, “PEG Modulated Release of Etanidazole from Implantable PLGA/PDLA Discs,” Biomaterials, Vol. 17, 2002, pp. 3555-3566. doi:10.1016/S0142-9612(02)00034-0
- D. Cohn and G. Lando, “Tailoring Lactide/Caprolactone Co-Oligomers as Tissue Adhesives,” Biomaterials, Vol. 25, No. 27, 2004, pp. 5875-5884. doi:10.1016/j.biomaterials.2004.01.040
- T. B. Reece, T. S. Maxey and I. L. Kron, “A Prospectus on Tissue Adhesives,” American Journal of Surgery, Vol. 182, No. 2, 2001, pp. 40S-44S. doi:10.1016/S0002-9610(01)00742-5
- Y. Baimark, N. Niamsa, N. Morakot, J. Threeprom and Y. Srisuwan, “Preparation and Morphology Study of Biodegradable Chitosan/Methoxy Poly(Ethylene Glycol)-bPoly (ε-Caprolactone) Nanocomposite Films,” International Journal of Polymer Analysis and Characterization, Vol. 12, No. 6, 2007, pp. 457-467.
- H. Korhonen, A. Helminen and J. V. Seppala, “Synthesis of Polylactides in the Presence of Co-Initiations with Different Numbers of Hydroxyl Groups,” Polymer, Vol. 42, No. 18, 2001, pp. 7541-7549. doi:10.1016/S0032-3861(01)00150-1
- H. M. Younes, E. B. Grimaldo and B. G. Amsden, “Synthesis, Characterization and in Vitro Degradation of a Biodegradable Elastomer,” Biomaterials, Vol. 25, No. 22, 2004, pp. 5261-5269. doi:10.1016/j.biomaterials.2003.12.024
- P. Perugini, I. Genta, B. Conti, T. Modena and F. Pavanetto, “Periodontal Delivery of Ipriflavone: New Chitosan/PLGA Film Delivery System for a Lipophilic Drug,” International Journal of Pharmaceutics, Vol. 252, No. 1-2, 2003, pp. 1-9. doi:10.1016/S0378-5173(02)00602-6
- A. L. Pataro, C. F. Franco, V. R. Santos, M. E. Cortes and R. D. Sinisterra, “Surface Effects and Desorption of Tetracycline Supramolecular Complex on Bovine Dentine,” Biomaterials, Vol. 24, No. 6, 2003, pp. 1075-1080. doi:10.1016/S0142-9612(02)00403-9
- A. Budhian, S. J. Siegel and K. I. Winey, “Controlling the in Vitro Release Profiles for a System of Haloperidol-Loaded PLGA Nanoparticles,” International Journal of Pharmaceutics, Vol. 346, No. 1-2, 2008, pp. 151-159. doi:10.1016/j.ijpharm.2007.06.011
NOTES
*Corresponding author.