Computational Chemistry
Vol.05 No.03(2017), Article ID:77975,16 pages
10.4236/cc.2017.53010
Theoretical Study of the Reaction of (2, 2)-Dichloro (Ethyl) Arylphosphine with Bis (2, 2)-Dichloro (Ethyl) Arylphosphine by Hydrophosphination Regioselective by the DFT Method
Kouadio Valery Bohoussou1, Anoubilé Benié2, Mamadou Guy-Richard Koné1, Affi Baudelaire Kakou2, Kafoumba Bamba1, Nahossé Ziao1
1Laboratoire de Thermodynamique et de Physico-Chimie du Milieu, UFR SFA, Université Nangui Abrogoua, Abidjan, République de Côte-d’Ivoire
2Laboratoire de Chimie Bio-Organique et de Substances Naturelles (LCBOSN), UFR SFA, Université Nangui Abrogoua, Abidjan, République de Côte-d’Ivoire

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: April 10, 2017; Accepted: July 24, 2017; Published: July 27, 2017
ABSTRACT
For this work, we have selected two reactions for the formation of (2,2)- dichloro (ethyl) Arylphosphine and bis (2,2)-dichloro(ethyl)arylphosphine compounds by hydrophosphination. Global and local reactivity parameters, thermodynamic parameters of reactions, Transition states, the Fukui function, the local softness, the local electrophility index, and nucleophility index, Natural population analyses (NPA) and Mulliken (MK) were calculated with DFT method at B3LYP/6-311+G(d, p) level. The analysis of potential energy surfaces and the nature of the reaction mechanism have been determined. The various results obtained revealed that the addition of Arylphosphine is regiospecific. The phenylphosphine is more stable than the thiophenylpho- sphine. The theoretical results are consistent with experience.
Keywords:
Hydrophosphination, Phosphine, HOMO, LUMO, Fukui Index, Transition State

1. Introduction
Organophosphorus compounds have enormous potentialities in various domains, including agriculture [1] , medicine [2] [3] , pharmacy, polymer sciences [4] , asymmetric catalysis [5] [6] [7] [8] [9] , chemical and agro-food industries, but their strong odor, their toxicity, their great sensitivity to air and their great instability make these compounds difficult to manipulate. Predicting their stability is therefore a major challenge for the synthesis of new optically active compounds. It is to this concern that the theoretical study of the reaction of hydrophosphination of free phosphines comes to answer. The aim of this work is to determine by means of theoretical chemistry the transition states and the reaction mechanism of two free phosphines synthesized by Benié et al. [10] . Experimentally, these authors found that the addition of primary phosphine to 1, 1-dichloroethylene is regioselective [10] (Anti-Markovnikov).
2. Computational Details
2.1 Calculation Level
2.2. Thermodynamic Reaction Parameters
The knowledge of the variations of the energy contributions to the internal energy at 0 K and at 298 K between the products and the reactants contributes to the energy characterization of a chemical reaction (Figure 1). For a given energy parameter X, its variation is determined according to relation:
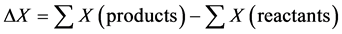 (1)
(1)
The variation of the internal energy at 298 K, constitutes a sum of the different contributions Electronic, translation, rotation, vibration and internal energy at 0K according to the relation:
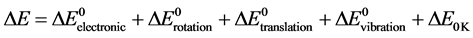 (2)
(2)
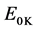 commonly referred to as Zero Point Vibrational Energy (ZPVE) translates the vibrational energy at the zero point induced by the 3N − 6 (respectively 3N − 5) normal vibration modes of frequency ni of the N cores of a non-Linear (or of a linear molecule) to 0 K. It is defined by the following relation:
commonly referred to as Zero Point Vibrational Energy (ZPVE) translates the vibrational energy at the zero point induced by the 3N − 6 (respectively 3N − 5) normal vibration modes of frequency ni of the N cores of a non-Linear (or of a linear molecule) to 0 K. It is defined by the following relation:
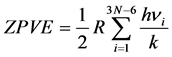 (3)
(3)
k the Boltzmann constant; h the Planck constant; R the perfect gas constant). As for the contributions of rotation and translation, they are derived from the approximation of perfect gases by the relation:
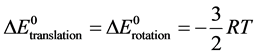 (4)
(4)
To obtain the corresponding energy at 298 K, it is necessary to take into account the additional energy due to the population of the vibration levels during the temperature rise from 0 to 298 K, defined by the relation:
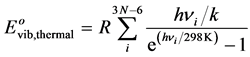 (5)
(5)
It follows that the internal energy variation at 298 K can be written as:
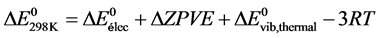
From this relationship, the enthalpy of reaction at 298 K is deduced. It corresponds to the variation of the corrected internal energy of the term Δ(PV), either ΔnRT (Δn being the change in the number of gaseous moles during the reaction):
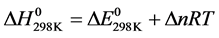 (7)
(7)
The entropy contributions of translation, rotation and vibration of a given species to 298 K are grouped in the total entropy term S. The entropy of the reaction is determined according to the relation:
 (8)
(8)
The Gibbs energy at 298 K, related to the reaction, is simply obtained by the relation:
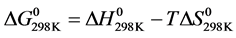 (9)
(9)
2.3. Conceptual DFT Reactivity Parameters
2.3.1. Global Reactivity Descriptors
According to Parr, the electronic chemical potential (μ) can be defined from the Lagrange multiplier [13] . This definition is exactly the same deduced by Pearson.
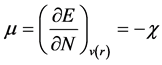 (10)
(10)
The electronic chemical potential μ can be calculated from the energies of the frontier molecular orbitals EHOMO and ELUMO as follows
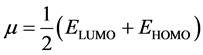 (11)
(11)
Other parameters of global reactivity called global hardness (η) and overall softness (S) were introduced from the variation of energy from one stationary state to another. These parameters are given by:
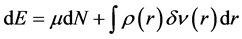 (12)
(12)
With µ: chemical potential, ρ(r): electron density, n(r): external potential of the system.
The first partial derivative of μ with respect to N (the total number of electrons) is defined as the overall hardness η of the system [15] [16] . It is related to the quantity S which is the global softness of the system.
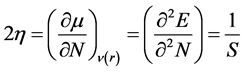 (13)
(13)
The global hardness η can also be calculated from the energies of the frontier molecular orbitals EHOMO and ELUMO as follows
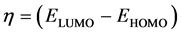 (14)
(14)
From the chemical hardness, a further parameter of reactivity is determined in order to define the energy stability due to the charge transfer. Global electrophilicity index (ω) [17] which is linked to the chemical potential by the following relation
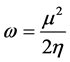 (15)
(15)
It should be noted that the nucleophilic index cannot be defined by a variational procedure, because there is no molecular electronic stabilization along the subtraction of the electron density of a molecule. In lack of a nucleophilic descriptor, Domingo et al. [18] proposed that the hypothesis that a weakly electrophilic molecule is systematically highly nucleophilic is true only for simple molecules. High values of nucleophilic correspond to low values of ionization potential and vice versa. Domingo et al. used the energy of the Highest Occupied Molecular Orbital (EHOMO) obtained by the Kohn-Sham method [18] . The empirical (relative) nucleophilic index (N) [19] is defined as follows:
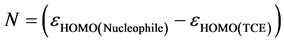 (16)
(16)
 : Energy of tetracyanoethylene (TCE)
: Energy of tetracyanoethylene (TCE)
Here, tetracyanoethylene is taken as the reference in the nucleophilicity scale, because it presents lowest HOMO energy.
The maximum charge transfer ( ) represents the maximum load proportion a system can acquire from its environment [20] . It is defined as follows:
) represents the maximum load proportion a system can acquire from its environment [20] . It is defined as follows:
 (17)
(17)
Electrophilicity gap (Δω) between the reactants makes it possible to predict the polarity of the reaction [21] . This descriptor is defined by:
 (18)
(18)
2.3.2. Local Reactivity Descriptors
Fukui functions  corresponding to the site k of a molecule is defined as the first derivative of the electronic density
corresponding to the site k of a molecule is defined as the first derivative of the electronic density  of a system with respect to the number of electrons N to a constant external potential
of a system with respect to the number of electrons N to a constant external potential  [22] .
[22] .
 (19)
(19)
The condensed form of Fukui functions in a molecule with N electrons has been proposed by Yang et al. [23] by:
 for nucleophilic attack (20)
for nucleophilic attack (20)
 for electrophilic attack (21)
for electrophilic attack (21)
 for radical attack (22)
for radical attack (22)
with
 : Electronic population of the atom k in the neutral molecule
: Electronic population of the atom k in the neutral molecule
 : Electronic population of the k atom in the anionic molecule
: Electronic population of the k atom in the anionic molecule
 : Electron population of the k atom in the cationic molecule
: Electron population of the k atom in the cationic molecule
The product of the global electrophilic index ω and the electrophilic Fukui index  make it possible to identify the most electrophilic site. This site is identified by the determination of the local electrophilic index (
make it possible to identify the most electrophilic site. This site is identified by the determination of the local electrophilic index ( ) [24] , defined as:
) [24] , defined as:
 (23)
(23)
Similarly, the most nucleophilic site can be identified by the local nucleophilic index,  [25] ; Defined as the product of the global nucleophilic index N and the nucleophilic Fukui index
[25] ; Defined as the product of the global nucleophilic index N and the nucleophilic Fukui index .
.
 (24)
(24)
According to Domingo et al., The local electrophile  and local nucleophilic index
and local nucleophilic index  are reliable descriptors for the prediction of the most favorable nucleophilic-electrophilic interaction for the formation of a chemical bond between two atoms [26] . This chemical bond takes place between the nucleophilic site (characterized by the highest
are reliable descriptors for the prediction of the most favorable nucleophilic-electrophilic interaction for the formation of a chemical bond between two atoms [26] . This chemical bond takes place between the nucleophilic site (characterized by the highest  value) of the nucleophilic molecule and the electrophilic site (characterized by the greater
value) of the nucleophilic molecule and the electrophilic site (characterized by the greater  value) of the electrophilic molecule.
value) of the electrophilic molecule.
The condensed Fukui Functions  and the global softness S also make it possible to determine the local softness
and the global softness S also make it possible to determine the local softness  [27] , defined by:
[27] , defined by:
 (25)
(25)
The condensed local softnesses  can be calculated from the following expressions:
can be calculated from the following expressions:
 (26)
(26)
where +, −, and 0 signs show nucleophilic, electrophilic, and radical attack, respectively.
According to the rule of Gazquez-Mendez [28] , Two chemical species interact through the atoms having equal or neighboring softnesses.
3. Results and Discussion
3.1. Thermodynamic Parameters
The standard internal energy ( ), the standard entropy (
), the standard entropy ( ), the standard enthalpy (
), the standard enthalpy ( ) and the standard free enthalpy (
) and the standard free enthalpy ( ) characterizing the hydrophosphination reaction of the primary phosphines (1a, 1b) on dichloroethylene (R2) are summarized in Table 1.
) characterizing the hydrophosphination reaction of the primary phosphines (1a, 1b) on dichloroethylene (R2) are summarized in Table 1.

Table 1. Thermodynamic parameters characterizing the hydrophosphination reaction of compounds 1a and 1b on R2 calculated at B3LYP 6-311+G(d, p).
All the values of the standard thermodynamic reaction parameters of the molecules are negative. These negative values of enthalpy and free enthalpy, respectively, translate an exothermic and spontaneous reaction under the conditions of the study. Negative values of internal energy reflect the formation and existence of products 2a, 2b, 3a and 3b. As far as entropy is concerned, all negative values reflect a decrease in disorder. Considering the free enthalpies of reaction, the order of decreasing stability of the products is deduced as follows: the tertiary phosphines 3a and 3b formed are more stable than the secondary phosphines 2a and 2b either (3a) more stable than (2a) and (3b) more stable than (2b).
3.2. Prediction of Electrophilic and Nucleophilic Character of Reactant
The values of the global descriptors of reactivity of reactants 1a, 1b and R2 are given in Table 2 below.
Table 2. Global descriptors of chemical reactivity of reactants 1a, 1b and R2 at B3LYP/ 6-311 +G(d, p).
EHOMO(TCE) = −9489 eV at Level B3LYP/6-311+ G(d, p).
The analysis of the chemical reactivity parameters Table 2 shows that, the chemical potential μ of the compound 1a (μ = −3.964 eV) is found on an energy level higher than that of the compound R2 (μ = −4.244). This observation shows that the transfer of electrons will take place from compound 1a to compound R2. Regarding the nucleophilic index, compounds 1a (N = 2.525) and 1b (N = 2.744) have values significantly greater than compound R2 (N = 1.992). This means that the compounds 1a and 1b are nucleophiles while the compound R2 is an electrophile. This trend is confirmed by the analysis of the electrophilic index values (ω) with the larger values for the compound R2. The hardness η shows that the free phosphines 1a and 1b having 6.000 eV and 5.645 eV values respectively are lower than that of dichloroethylene R2 (6.507 eV). This means that the free phosphines 1a and 1b maintains their electrons weak in their environment, unlike the dichloroethylene R2 which maintains them in its environment. Therefore electron transfer takes place from the phosphine to the dichloroethylene.
Global electrophilicity gap parameter of the two reactants (1a/R2 and 1b/R2) is less than 1. This indicates a weak polar character for this addition. The last parameter studied is the HOMO-LUMO energy gap of the reactants 1a/R2 and 1b/R2 (Figure 2).
3.3. Prediction of Local Descriptors of Reactant
3.3.1. Prediction Using the Domingo Model
The values of the local reactivity descriptors, in this case the Fukui function ( ,
, ), the local softness (
), the local softness ( ,
, ), the local electrophile power (
), the local electrophile power ( ), and the dual descriptors (
), and the dual descriptors ( ) of the reactivity have been determined. These values of the local descriptors on the atoms P1, P2, C1 and C2 of the reactants 1a, 1b and R2 calculated according to the model of Domingo et al. With the Natural Population Atomic (NPA) [29] [30] [31] and Mulliken (MK) [31] approaches at B3LYP/6-311+G(d, p) are reported in Table 3.
) of the reactivity have been determined. These values of the local descriptors on the atoms P1, P2, C1 and C2 of the reactants 1a, 1b and R2 calculated according to the model of Domingo et al. With the Natural Population Atomic (NPA) [29] [30] [31] and Mulliken (MK) [31] approaches at B3LYP/6-311+G(d, p) are reported in Table 3.
On analysis of the values in Table 3, phosphines 1a and 1b have the highest values of the local nucleophilic indices . Similarly, the carbon C1 of the compound R2 has the highest value of the local electrophilic index (
. Similarly, the carbon C1 of the compound R2 has the highest value of the local electrophilic index ( ). This shows that the most favored interaction takes place between the P1 atom of the compound 1a and the C1 atom of the compound R2 for the first reaction, and between the P9 and C1 atoms for the second reaction. Therefore, the formation of experimentally observed P1-C1 and P2-C1 bonds are correctly predicted by the Domingo model with the Mulliken and NPA approaches.
). This shows that the most favored interaction takes place between the P1 atom of the compound 1a and the C1 atom of the compound R2 for the first reaction, and between the P9 and C1 atoms for the second reaction. Therefore, the formation of experimentally observed P1-C1 and P2-C1 bonds are correctly predicted by the Domingo model with the Mulliken and NPA approaches.
Table 3. Local reactivity descriptors on the P1, P2, C1 and C2 atoms of reactants 1a, 1b and R2 using NPA and Mulliken population analyzes at B3LYP/6-311 + G(d, p).
3.3.2. Prediction Using the Gazquez-Mendez Model
The prediction according to the Gazquez-Mendez model presents values of the Fukui function ( ,
, ), local softness
), local softness  for reactants R2 and local softness
for reactants R2 and local softness  for reactants 1a or 1b. These values of the local descriptors on the atoms P1, P2, C1 and C2 of the reactants 1a, 1b and R2 were calculated according to the Gazquez-Mendez model with the NPA population analyzes and MK at the B3LYP 6-311+G level (d, p) are given in Table 4.
for reactants 1a or 1b. These values of the local descriptors on the atoms P1, P2, C1 and C2 of the reactants 1a, 1b and R2 were calculated according to the Gazquez-Mendez model with the NPA population analyzes and MK at the B3LYP 6-311+G level (d, p) are given in Table 4.
Examination of the values in Table 4 indicates that the phosphines 1a, 1b and dichloroethylene R2 have similar values of local softnesses ( ,
, ) respectively on P1, P2 and C1 atoms by the approach of Mulliken. This observation shows
) respectively on P1, P2 and C1 atoms by the approach of Mulliken. This observation shows
Table 4. Values of Fukui Functions ( ,
, ), local softnesses
), local softnesses  for reactants R2 and local softness
for reactants R2 and local softness  for reactants 1a and 1b calculated by NPA, MK.
for reactants 1a and 1b calculated by NPA, MK.
that the most favored interaction takes place between the P1 atom of the compound 1a and the C1 atom of the dichloroethylene for the first reaction and the P2 atom of the compound 1b and the C1 atom of the dichloroethylene for the second reaction. Also in Table 4, with the NPA approach, the values of the local softnesses  of the phosphines 1a and 1b on the atoms P1 and P2 are closer to the values of the local softnesses
of the phosphines 1a and 1b on the atoms P1 and P2 are closer to the values of the local softnesses  The C1 atom of the compound R2 compared to the C2 atom, which also confirms the interaction between the phosphors of the various phosphines and the least substituted carbon.
The C1 atom of the compound R2 compared to the C2 atom, which also confirms the interaction between the phosphors of the various phosphines and the least substituted carbon.
3.4. Potential Energy Surfaces and Prediction of the Reaction Mechanism
3.4.1. In the Gas
In order to elucidate the reaction mechanism and compare the stability of the formed phosphines, the values of the reactant energies, the energies of the products, the energies of the transitions states and the activating energies are presented in Table 5 below.
Table 5. The energies of the transition states (ETS), activation (Ea) and reactants corresponding to the formation of products 2a and 2b calculated at the B3LYP/6-31G(d, p).
From Table 5, it can be seen that the activation energy found by the reaction pathway give a value of 0.03765 kcal/mol. This value is inferior to what has been found by the reaction pathway through 2b (29.4925 kcal/mol). The phosphine 2a is more stable than that of 2b.
In order to show that the transition states (TS) are well connected to minima (reactants and products), the calculation of the Intrinsic Reactions Coordinates (IRC) and the curves of the reaction pathway, E = f (RC) are shown in Figures 3-5.

Figure 3. IRC curve of reaction pathway 2a.

Figure 4. IRC curve of reaction pathway 2b.
The evolution of the IRC curve (Figure 4), Indicates that the energy of the product obtained (−1892.770 Hartree) is greater than that of the reactant (−1892.772 Hartree) with thiophenyl as radical. This means that the compound (2b) obtained is kinetically unstable. The product 2a is therefore more stable than the product 2b, under the same conditions.
Figure 3 shows that the phosphorus carbon (P-C) bond is established in the transition state on the most hydrogenated C1 carbon and the hydrogen of the phosphorus then on the C2 carbon bonded to the chlorine. Similarly, the phosphorus carbon bond is established first on the most hydrogenated carbon during the formation of the product 3a. In the transition state, the hydrogen carbon bond is not yet established (Figure 5).

Figure 5. IRC curve of reaction pathway 3a.
Reaction pathway therefore makes it possible to propose the following reaction mechanism Figure 6.

Figure 6. Proposed Mechanism for the Hydrophosphination of free phosphine with dichloroethylene.
The products 3a and 3b have been obtained after the formation of the products 2a and 2b.
Thus, during the addition of 1a to R2, respectively 1b to R2, two reactions occur in succession:

This explains the stability of the products 3a and 3b obtained experimentally and the fact that they are major products [10] .
Products 3a and 3b are obtained in an excess of reactant (R2), unlike product 2a and 2b which require less reactant.
3.4.2. In the Solvent
The influence of the solvent on the getting of the product 2a was envisaged at B3LYP/6-31G (d, p). The results are shown in Table 6 below.
The study of the addition reaction envisaged in the solvents gives satisfactory results at the level of theory used. The activation barrier (Ea = 2.51 kcal / mol) is the lowest when the diethylene is used as a solvent. The diethylene favors the obtaining of a product kinetically more stable product. Consequently, the product 2a can be obtained optimally in diethylene. These theoretical predictions are in agreement with the experimental studies carried out (Figure 7).
4. Conclusion
The reaction mechanism and the transition states of the two free phosphines were studied by the DFT method. Electrophilic and nucleophilic character, electrophilic and nucleophilic local indices, Fukui Functions, condensed local softness, thermodynamic quantities of the addition reaction of two arylphosphines on dichloroethylene, localization of transition states, atomic electronic populations and reactivity indices calculated by means of natural population analyses (NPA) and Mulliken (MK), and analysis of the potential energy surface have allowed us to conclude that: the most favored interaction takes place between the phosphorus atom (P1) of the arylphosphine and the most hydrogenated carbon (C1) of the dichloroethylene.
The majority product is obtained after a successive addition of the Arylphosphine to the dichloroethylene and then an addition of the product obtained (2, 2) dichloro (ethyl) Arylphosphine) to the dichloroethylene. 2, 2-dichloro (ethyl) phenylphosphine can be obtained after a single step in the solvent (diethylether). The addition reaction of Arylphosphine on dichloroethylene is regioselective “anti-Markovnikov”. Phenylphosphine is more stable than thiophenylphosphine. The total energy values of the reaction are negative, which implies that the reaction is exothermic.
Cite this paper
Bohoussou, K.V., Benié, A., Koné, M.G.-R., Kakou, A.B., Bamba, K. and Ziao, N. (2017) Theoretical Study of the Reaction of (2, 2)-Dichloro (Ethyl) Arylphosphine with Bis (2, 2)- Dichloro (Ethyl) Arylphosphine by Hydro- phosphination Regioselective by the DFT Method. Computational Chemistry, 5, 113- 128. https://doi.org/10.4236/cc.2017.53010
References
- 1. Sharma, S.B., Sayyed, R.Z., Trivedi, M.H. and Gobi, T.A. (2013) Phosphate Solubilizing Microbes: Sustainable Approach for Managing Phosphorus Deficiency in Agricultural Soils. Springerplus, 2, 587.
https://doi.org/10.1186/2193-1801-2-587 - 2. Nigam, S., Burke, B.P., Davies, L.H., Domarkas, J., Wallis, J., Waddell, F.P.G., Waby, J.S., Benoit, D.M., Seymour, A., Cawthorne, C., Higham, L.J. and Archibald, S.J. (2016) Structurally Optimised BODIPY Derivatives for Imaging of Mitochondrial Dysfunction in Cancer and Heart Cells. Chemical Communications (Cambridge), 52, 7114-7117.
https://doi.org/10.1039/C5CC08325G - 3. Katti, K.V., Pillarsetty, N. and Raghuraman, K (2003) New Vistas in Chemistry and Applications of Primary Phosphines. Topics in Current Chemistry, 229, 121-141.
https://doi.org/10.1007/b11153 - 4. Clark, T.J., Rodezno, J.M., Clendenning, S.B., Aouba, S., Brodersen, P.M., Lough, A.J., Ruda, H.E. and Manners, I. (2005) Rhodium Dehydrocoupling of Fluorinated Phosphine-Borane Adducts: Synthesis, Characterization, and Properties of Cyclic and Polymeric Phosphinoboranes with Electron-Withdrawing Substituents at Phosphorus. Chemistry: A European Journal, 11, 4526-4534.
https://doi.org/10.1002/chem.200401296 - 5. Xiao, Y., Sun, Z., Guo, H. and Kwon, O. (2014) Chiral Phosphines in Nucleophilic Organocatalysis. Beilstein Journal of Organic Chemistry, 10, 2089-2121.
- 6. Clarke, T.P. and Landis, C.R. (2004) Recent Developments in Chiral Phospholane Chemistry. Tetrahedron Asymmetry, 15, 2123-2137.
https://doi.org/10.1016/j.tetasy.2004.06.025 - 7. Hoge, G. and Samas, B. (2004) Application of P-Chirogenic Bisphospholane Ligands to Rhodium Catalyzed Asymmetric Hydrogenation of α- and β-Acetamido Dehydroamino Acid Derivatives. Tetrahedron: Asymmetry, 15, 2155-2157.
https://doi.org/10.1016/j.tetasy.2004.04.041 - 8. Brauer, D.J., Kottsieper, K.W., Roβenbach, S. and Stelzer, O. (2003) Novel P,N Ligands Derived from (R)- and (S)-1-Phenylethylamine with (2R,5R)-2,5-Dimethylphospholanyl Groups (DuPHAMIN) for Asymmetric Catalysis. European Journal of Inorganic Chemistry, 2003, 1748-1755.
https://doi.org/10.1002/ejic.200200388 - 9. Herrbach, A., Marinetti, A., Baudoin, O., Guénard, D. and Gueritte, F.J. (2003) Asymmetric Synthesis of an Axially Chiral Antimitotic Biaryl via an Atropo-Enantioselective Suzuki Cross-Coupling. Journal of Organic Chemistry, 68, 4897-4905.
- 10. Benié, A., Dénis, J.M. and Békro, Y.A. (2010) Synthesis of Free and Complexes 2,2 Dichloro(ethyl)Arylphosphine and bis(2,2)Dichloro(ethyl)Arylphosphine by Regioselective Hydrophosphination. European Journal of Scientific Research, 47, 164-168.
http://www.lablcbosn.com/wp-content/uploads/2015/09/ejsr_47_2_01.pdf - 11. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery, Jr., J.A., Vreven, T., Kudin, K.N., Burant, J.C., Millam, J.M., Iyengar, S.S., Tomasi, J., Barone, V., Mennucci, B., Cossi, M., Scalmani, G., Rega, N., Petersson, G.A., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li, X., Knox, J.E., Hratchian, H.P., Cross, J.B., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Ayala, P.Y., Morokuma, K., Voth, G.A., Salvador, P., Dannenberg, J.J., Zakrzewski, V.G., Dapprich, S., Daniels, A.D., Strain, M.C., Farkas, O., Malick, D.K., Rabuck, A.D., Raghavachari, K., Foresman, J.B., Ortiz, J.V., Cui, Q., Baboul, A.G., Clifford, S., Cioslowski, J., Stefanov, B.B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Martin, R.L., Fox, D.J., Keith, T., Al-Laham, M.A., Peng, C.Y., Nanayakkara, A., Challacombe, M., Gill, P.M.W., Johnson, B., Chen, W., Wong, M.W., Gonzalez, C. and Pople, J.A. (2004) Gaussian 03, Revision C. 01. Gaussian Inc., Wallingford.
- 12. Becke, A.D. (1993) A New Mixing of Hartree-Fock and Local Density-Functional Theories. Journal of Chemical Physics, 98, 1372-1377.
https://doi.org/10.1063/1.464304 - 13. Parr, R.G. and Yang, W. (1989) Density-Functional Theory of Atoms and Molecules. Oxford University Press, New York, Oxford.
- 14. De Castro, E.V.R. and Jorge, F.E. (1998) Accurate Universal Gaussian Basis Set for All Atoms of the Periodic Table. Journal of Chemical Physics, 108, 5225-5229.
https://doi.org/10.1063/1.475959 - 15. Parr, R.G. and Pearson, R.G. (1983) Absolute Hardness: Companion Parameter to Absolute Electronegativity. Journal of the American Chemical Society, 105, 7512-7516.
https://doi.org/10.1021/ja00364a005 - 16. Yang, W. and Parr, R.G. (1985) Hardness, Softness, and the Fukui Function in the Electronic Theory of Metals and Catalysis. Proceedings of the National Academy of Sciences, 82, 6723-6726.
https://doi.org/10.1073/pnas.82.20.6723 - 17. Chattaraj, P.K., Sarka, U.R. and Roy, D.R. (2006) Electrophilicity Index. Chemical Reviews, 106, 2065-2091.
https://doi.org/10.1021/cr040109f - 18. Domingo, L.R., Aurell, M.J., Perez, P. and Contreras, R. (2002) Quantitative Characterization of the Global Electrophilicity Power of Common Diene/Dienophile Pairs in Diels-Ald Reactions. Tetrahedron, 58, 4417-4423.
- 19. Domingo, L.R., Chamorro, E. and Pérez, P. (2008) Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. The Journal of Organic Chemistry, 73, 4615-4624.
https://doi.org/10.1021/jo800572a - 20. Parr, R.G., von Szentpaly, L. and Liu, S. (1999) Electrophilicity Index. Journal of the American Chemical Society, 121, 1922-1924.
https://doi.org/10.1021/ja983494x - 21. Delgado, J.S., Aizman, A., Domingo, L.R. and Contreras, R. (2010) Understanding the Influence of Lewis Acids in the Regioselectivity of the Diels-Alder Reactions of 2-Methoxy-5 Methyl 1,4 Benzoquinone: A DFT Study. Chemical Physics Letters, 449, 272.
- 22. Parr, R.G. and Yang, W. (1984) Perspective on Density Functional Approach to the Frontier-Electron Theory of Chemical Reactivit. Journal of the American Chemical Society, 106, 4049-4050.
- 23. Domingo, L.R. and Saez, J.A. (2009) Understanding the Mechanism of Polar Diels-Alder Reactions. Organic and Biomolecular Chemistry, 7, 3576-3583.
https://doi.org/10.1039/b909611f - 24. Toledo, O.R. and Contreras, R. (2014) Philicity and Fugality Scales for Organic Reactions. Advances in Chemistry, 2014, 1-13.
- 25. Reed, A.E. and Weinhold, F. (1983) Natural Atomic Orbitals and Natural Population Analysis. Journal of Chemical Physics, 78, 4066-4073.
https://doi.org/10.1063/1.445134 - 26. Domingo, L.R., Aurell, M.J., Perez, P. and Contreras, R. (2000) Quantitative Characterization of the Local Electrophilicity of Organic Molecules. Understanding the Regioselectivity on Diels-Alder Reactions. Journal of Chemical Physics, 106, 6871-6875.
https://doi.org/10.1021/jp020715j - 27. Yan, W.G. and Parr, R.G. (2000) Hardness, Softness, and the Fukui Function in the Electronic Theory of Metals and Catalysis. Proceedings of the National Academy of Sciences, 82, 6723-6726.
- 28. Mendez, F. and Gazquez, J.L. (1994) Chemical Reactivity of Enolate Ions: The Local Hard and Soft Acids and Bases Principle Viewpoint. Journal of the American Chemical Society, 116, 9298-9301.
https://doi.org/10.1021/ja00099a055 - 29. Reed, A. and Weinhold, E.F. (1983) Natural Atomic Orbitals and Natural Population Analysis. Journal of Chemical Physics, 78, 4066-4073.
- 30. Reed, A.E., Weinstock, R.B. and Weinhold, F. (1985) Natural Atomic Orbitals and Natural Population Analysis. Journal of Chemical Physics, 83, 735-746.
https://doi.org/10.1063/1.449486 - 31. Besler, B.H., Merz, K.M.J. and Kollman, P.A. (1990) Atomic Charges Derived from Semi Empirical Methods. Journal of Computational Chemistry, 11, 431-439.
https://doi.org/10.1002/jcc.540110404