Advances in Biological Chemistry
Vol.3 No.2(2013), Article ID:30757,7 pages DOI:10.4236/abc.2013.32022
Purification and characterization of glyceraldehyde-3-phosphate dehydrogenase from saline strain Idiomarina loihiensis
![]()
1Laboratoire de Physiologie et Génétique Moléculaire, Faculté des Sciences Aïn Chock, Université Hassan II-Aïn Chock, Casablanca, Morocco
2UR Étude et Gestion des Environnements Urbains et Côtiers, LARSEN, Université de Sfax, École Nationale d’Ingénieurs de Sfax, Sfax, Tunisia
Email: *mardadilham@yahoo.fr, a.soukri@hotmail.com
Copyright © 2013 Ilham Mardad et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 9 January 2013; revised 4 March 2013; accepted 12 April 2013
Keywords: Glyceraldehyde-3-Phosphate; Dehydrogenase; Idiomarina loihiensis; Purification; NAD+; Kinetics; Saline Strain
ABSTRACT
Idiomarina loihiensis was isolated from the salt works in Sfax (Tunisia), until now, the characterization of the GAPDH phosphorylante was never studied. Here, we report the isolation and the biochemical characterization of glyceralehyde-3-phosphate dehydrogenase (GAPDH) from I. loihiensis saline’s bacteria on the basis of the apparent native and subunit molecular weights, physico-chemical and kinetic characterizations. The purification method consisted of two steps, ammonium sulfate fractionation followed by one chromatographic step, namely dye-affinity on Blue Sepharose CL-6B. Polyclonal antibodies against the purified enzyme were used to recognize the I. loihiensis GAPDH by Western blotting. The optimum pH of the purified enzyme was 8.5. Studies on the effect of temperatures revealed an enzyme increasing activity of about 45˚C. The molecular weight of the purified enzyme was 36 kDa determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Non-denaturing polyacrylamide gels yield a molecular weight of 147 kDa. The Michaelis constants for NAD+ and D-glyceraldehyde-3-phosphate estimated was 19 μM and 3.1 μM, respectively. The maximal velocity of the purified enzyme was estimated to be 2.06 U/mg, approximately 6-fold increase in specific activity and a final yield of approximately 32.5%. The physicochemical properties of this GAPDH, being characterized, could be used in further studies.
1. INTRODUCTION
Idiomarina loihiensis (lo.i.hi.en’sis.N.L.fem.agj.loihiensis) originating from Lōʻihi, the site of isolation of the type strain is a deep-sea living Gram-negative, 0.35 µm wide and 0.7 - 1.0 µm long gamma-proteobacterium, which may occasionally rich up to 1.8 µm in length. Cells are motile by a single polar or subpolar flagellum and the genome comprises a single chromosome of 2,839,318 base pairs.
Idiomarina loihiensis was isolated from the salt works in Sfax (Tunisia). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is an enzyme involved in central pathways of carbon metabolism. The most common form of GAPDH is the NAD+-dependent enzyme (EC 1.2.1.12) found in all organisms so far studied and located in the cytoplasm. This enzyme plays a role in the EmbdenMeyerhoff pathway not only in glycolysis but also in gluconeogenesis [1].
Many new additional functions for GAPDH have been described. These novoties include its requirement for transcriptional control of histone gene expression, its essential function in nuclear membrane fusion, its necessity for the recognition of fraudulently incorporated nucleotides in DNA, and its mandatory participation in the maintenance of telomere structure [2,3]. These new functions require GAPDH association with a series of multi-enzyme complexes. Although other proteins in those complexes are variable, GAPDH remains the single constant protein in each structure.
This enzyme is broadly distributed in nature in a variety of organisms ranging from bacterial cells to human tissues [4,5]. It is found for the most part in the cytosol, mitochondria, and chloroplasts in plants. Generally, these molecules are all tetramers composed of apparently identical subunits [6-8] and have molecular weights ranging from 120 to 200 kDa [6,9].
In the cell, the study of proteins associated with phosphate management is one of the most important fields of research for comprehension of the phosphate metabolism in the bacteria. Among these proteins, glycreraldehyde- 3-phosphate dehydrogenase (GAPDH), the key enzyme of the cellular metabolism, was found at all the organizations study [1]. It fulfils a catabolic function allowing oxidative phosphorylation of glyceraldehyde-3-phosphate (G3P) in acid 1,3-diphosphoglyceric (1,3-DPG) and the reduction of NAD+ to NADH. During glycolysis, this reaction couples a dehydrogenation with a phosphorylation allowing the energy storage resulting from oxidation to later generate an ATP with the kinase of the 3-PG, an ATP. In this study, we report the isolation, the purification and the biochemical characterization of Idiomarina loihiensis GAPDH.
2. MATERIALS & METHODS
2.1. Strains and Culture Conditions
Isolation of saline’s bacteria:
The isolated bacteria from salt works, were cultivated in medium M2: NaCl, 98 g; KCl, 2 g; MgSO4, 7H2O, 1 g; CaCl2, 2H2O, 0.36 g; NaHCO3, 0.06 g; NaBr, 0.24 g; FeCl3, 6H2O, 1 g; Bactotryptone (Difco), 10 g; glucose, 1 g; agar, 20 g (per liter).
The pH was adjusted to 7.4. The plates were incubated at 37˚C aerobically in a salt saturated atmosphere. The isolated bacterial strains were cultivated and identified.
In the present study the purification of the GAPDH was done with the Idiomarina loihiensis which solubilizes the inorganic phosphate in the NBRIP medium. Idiomarina loihiensis was cultivated in medium LuriaBertani (LB) with NaCl and the enzyme was purified using traditional procedure [10,11] modified according to our conditions. All purification, steps were carried out at 4˚C.
2.2. Cell-Free Extract Preparation
Liquid culture cells were harvested by centrifugation at 15,000× g for 20 min at 4˚C. Cell pellets were washed twice in 25 mM Tris-HCl (pH 7.5) and resuspended in the same buffer supplemented with 2 mM EDTA and 10 mM β-mercaptoethanol). Cells were then disrupted by ultrasonic treatment in a chilled water bath using a Branson 25U sonifier at medium strength. The resulting suspension was centrifuged at 15,000× g for 45 min to obtain the cell-free extract.
2.3. GAPDH Activity Determination
Phosphorylating NAD+-dependent GAPDH activity was measured as described by Heina and Freimuller [12]. The reaction was started by the addition of 10 μg of cell-free extract to an assay mixture containing 50 mM Tricine buffer (pH 8.5), 1 mM NAD+, inorganic phosphate or arsenate (10 mM) in the reaction solution and 2 mM D-glyceraldehyde-3-phosphate at 25˚C. Absorbance at 340 nm was measured in a spectrophotometer (model BioMate 3, Thermo Electron Corporation, Madison WI 53711, USA).
2.4. GAPDH Purification
The crude extract was subjected to protein precipitation in the 60% - 88% (W/V) saturation range of ammonium sulphate. The final pellet was dissolved in a minimal volume of 25 mM Tris-HCl buffer (pH 7.5), containing 2 mM EDTA and 10 mM 2β-mercaptoethanol (buffer A). The protein solution was dialyzed twice against 2 l of the same buffer overnight.
The dialyzed enzyme preparation was applied to a Blue Sepharose CL-6B column (1 cm × 10 cm) equilibrated with 100 ml buffer A (approximately 10 times bed volumes).
The column was washed with 10 bed volumes of buffer A and 5 bed volumes of the same buffer adjusted to pH 9.5 (buffer B).
The enzyme was eluted with buffer B containing 10 mM NAD+ at a flow rate of 10 ml/h. Active fractions were collected, pooled and preserved in 50% glycerol (V/V) at −20˚C.
During enzyme purification, a coupled assay in which aldolase (1 U/ml) produced the stoichiometric breakage of D-fructose 1 - 6 biphosphate (2 mM) to dihydroxyacetone-phosphate and D-G3P the actual substrate of the oxidative reaction [13], was used.
2.5. Polyacrylamide Gel Electrophoresis
Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) was carried out as described by Laemmli [14] on 12% polyacrylamide slab gels containing 0.1% SDS. Gels were run on a miniature vertical slab gel unit (Hoefer Scientific Instruments, San Francisco, USA). After electrophoresis, the gels were stained with Coomassie Brilliant Blue R-250 at 0.2% (W/V) in the mixture of methanol/acetic acid/water (4:1:5, V/V/V) for 30 min at room temperature.
The apparent subunit molecular weight was determined by measuring and comparing relative mobility of the following pre-stained SDS-PAGE molecular weight standards (Broad Range Protein Molecular Weight Markers; Promega).
Determination of native molecular weight was carried out by electrophoresis on non-denaturing polyacrylamide slab gels (Bio-Rad, Hercules, USA) using the following protein standards: Apoferritin (443 kDa); Amylase (200 kDa); Alcohol dehydrogenase (150 kDa); Monomeric BSA (66 kDa); Carbonic anhydrase (29 kDa); and Lactalbumin (14.2 kDa). As described by the method of Hedrick and Smith [15], a calibration curve can be calculated from the relative mobility of standard proteins on non-denaturing polyacrylamide gels with different acrylamide concentrations (6%, 8%, 10%, and 12%, W/V).
By constructing the Ferguson plot [Log(Rf × 100) versus the concentration of polyacrylamide gels (%)], the resulting slopes versus the known molecular weights of standard native proteins allowed the determination of the native molecular weight of purified GAPDH.
2.6. Preparation of Polyclonal Antiserum
Polyclonal antiserum was raised in New Zealand White rabbits to GAPDH that had been purified from Idiomarina loihiensis. The purified protein (approximately 0.5 mg) was mixed 1:1 with Freund’s complete adjuvant. After 21 days, a sample of blood was collected, and a second dose of 500 µg of the protein was injected into rabbits in multiple places, as described by Vaitukaitis [16]. After one week, 50 ml of rabbit blood was collected and serum was separated after its coagulation overnight at 4˚C then its centrifugation. The obtained serum, containing monospecific anti-GAPDH polyclonal antibodies, was sampled and stored at −20˚C.
2.7. Western Blot Analysis
Proteins were separated by SDS-PAGE as described previously. Separated protein bands were electrophoretically transferred from the slab gel to a nitrocellulose membrane (Schleicher and Schuell, Keene, USA) using a BioRad Trans-Blot system. The transferred proteins were then visualized by pre-staining in 0.2% (W/V) Ponceau red in trichloroacetic acid.
The nitrocellulose membrane was then incubated for 1 h in blocking solution containing 5% (W/V) non-fat dry milk, 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.01% (W/V) NaN3, and 0.05% (V/V) Tween-20, followed by incubation with the anti-GAPDH antiserum (1:1000 dilution) as a primary antibody. Western blots were visualized by coupled immunoreaction with peroxidase-conjugated goat anti-rabbit immunoglobulin G (1:1000; Boehringer Mannheim, Hamburg, Germany) as a secondary antibody using 4-chloro-1-naphthol as a chromogenic substrate.
2.8. Kinetic Studies
For kinetic studies, initial velocities of the enzymatic reaction were carried out by varying the concentration of the substrates, D-G3P (from 0.04 to 10 mM) or NAD+ (from 0.02 to 2 mM). Values of the Michaelis constants (Km), dissociation constants (KD), and maximal velocity (Vmax) were calculated according to the method of Cleland [17]. One unit of enzyme activity was defined as the amount of enzyme that catalyses the formation of 1 μmol NADH/min under the conditions used. Protein concentrations were estimated by the method of Bradford [18] using bovine serum albumin (BSA) as a standard. Activity levels in cell-free extracts were expressed as specific activity (U/mg protein).
2.9. Determination of Optimal pH and Temperature for the Purified GAPDH Activity
To determine optimal pH, enzymatic activity was measured over a wide range of pH values (4 - 10), using a mixture of different buffers with different pKa (Tris, MES, HEPES, potassium phosphate at 50 mM, and sodium acetate at 180 mM) adjusted to the same ionic strength than the standard reaction mixture. Temperature effects were characterized by activation and denaturation processes. For activation, the tricine-NaOH buffer (50 mM; pH 8.5) was incubated for 10 min at temperatures from 20˚C to 80˚C using a thermostated cuvette holder connected to a refrigerated bath circulator. Then 2 mM NAD+, 200 mM sodium arsenate, and 10 μg purified GAPDH were added to the mixture. The reaction was started immediately by addition of 10 mM D-G3P.
For the denaturation, 10 μg of the purified GAPDH were incubated at temperatures from 20˚C to 80˚C for 10 min in the 50 mM tricine-NaOH buffer. Then 2 mM NAD+ and 200 mM sodium arsenate were added. The enzymatic activities were measured after 2 min incubation at 25˚C immediately started by addition of 10 mM D-G3P.
3. RESULTS
3.1. Enzyme Purification and Characterization
GAPDH was purified from a soluble protein fraction of the saline strain Idiomarina loihiensis crude extract, using a simple procedure involving only one chromatography step, namely dye affinity chromatography on Blue Sepharose. Table 1 summarizes a representative purification protocol. Approximately 1.8 U/mg proteins were obtained for the specific activity of the purified enzyme with a yield of 32.5% and an approximately 6-fold increase of purification.
SDS-PAGE analysis of the different fractions obtained during the purification procedure showed a progressive enrichment of a 36 kDa protein [Figure 1]. Only this protein band, with the same size as the GAPDH subunit

Figure 1. Purification of GAPDH from Idiomarina loihiensis detected by sodium dodecylsulfatepolyacrylamide gel electrophoresis (SDS-PAGE) (a) and Western blot analysis (b); Proteins at each purification step were resolved by SDS-PAGE and stained with Coomassie Brilliant Blue (a) or subjected to Western blot analysis (b) using polyclonal antibodies specific to Idiomarina GAPDH. Lane 2, crude extract (20 μg protein); Lane 3, protein preparation from the ammonium sulfate fractionation (66% - 88%) (20 μg); Lane 4, Blue Sepharose fractions pool (20 μg protein). The positions and apparent molecular weights of pre-stained standard proteins loaded in Lane 1 [Figure 1(a)] have been indicated. Bound antibody was located by coupled immunoreactions with peroxidase-conjugated goat antirabbit immunoglobulin G. The arrow indicates the band corresponding to the 36 kDa GAPDH.
Table 1. Purification of GAPDH from Idiomarina loihiensis.

monomer, was evidenced in the electrophoretically homogeneous final enzyme preparation [Figure 1(a), Lane 4].
Non-denaturing PAGE showed that the native molecular weight of the isolated protein was approximately of 147 kDa [Figure 2].
3.2. Kinetic Properties and Influence of pH and Temperature on Purified GAPDH Activity
Values of the Michaelis constants (Km), dissociation constants (KD) and maximal velocity for the oxidation of G3P and the reduction of NAD+ by the GAPDH were calculated. The KmNAD+, KmG3P and KDNAD+ values were estimated and showed a significant difference (Table 2). The Vmax of the purified protein was estimated to be 2.06 U/mg (Table 2). In comparison with the GAPDH from human erythrocytes, the KmNAD+, KmG3P and Vmax values were 17.8 mM, 205 mM and 4.3 U/mg respectively [19] and for Tetrahymena pyriformis, they were 5 mM, 150 mM and 5.6 U/mg respectively [20] and for Pleurodeles waltl, they were 60 mM, 27 mM and 33.2 U/mg [21].
Pre-incubation of Idiomarina GAPDH for 10 min at
Table 2. Kinetic constants (Km and Vmax) for oxidation reaction of GAPDH from Idiomarina loihiensis.

temperatures varying between 15˚C and 45˚C did not irreversibly affect the enzyme activity. However, thermal inactivation occurred above 50˚C and even at 80˚C, but no total activity loss was noted [Figure 3(a)]. Studies on the effect of temperature on enzyme activity revealed an optimal value around 45˚C.
The pH activity profile of purified GAPDH was determined in pH range from 4 to 10; the maximum of relative enzymatic activity is observed between pH 8 and 8.5 (Figure 3(b)).
4. DISCUSSION
Glycolysis is the main pathway for carbohydrate degradation in nearly all organisms. The generation of the final product of glycolysis from glucose, pyruvate, is completed by nine enzymatic steps, most of which function in the reverse direction during gluconeogenesis.

Figure 2. Determination of the native molecular weight of the purified Idiomarina loihiensis GAPDH by native gel electrophoresis. Proteins were electrophoresed on various polyacrylamide concentrations (6%, 8%, 10%, and 12%) under non-denaturing conditions. Molecular weight marker proteins were Apoferritin (443 kDa); Amylase (200 kDa); Alcohol dehydrogenase (150 kDa); Monomeric BSA (66 kDa); Carbonic anhydrase (29 kDa); and Lactalbumin (14.2 kDa). Relative mobility of proteins plotted as Log Rf (Relative mobility—100) versus gel concentration are indicated on inset. A plot of the obtained slopes versus molecular weight was linear and used to determine native Idiomarina loihiensis GAPDH molecular weight.
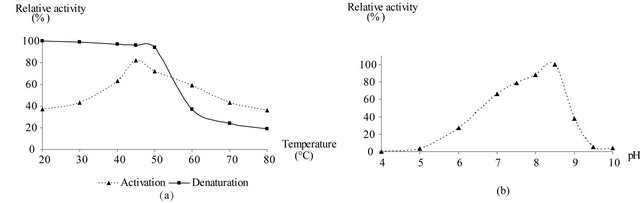
Figure 3. Determination of optimal temperature and pH for GAPDH of Idiomarina loihiensis. (a) Effect of the temperature on the purified GAPDH activity. This was followed by activation and denaturation processes at different temperatures (from 20˚C to 85˚C). (b) Relative activity of the purified GAPDH in the pH range 4.0 - 10.0 using a mixture of different buffers. The values represent the mean of three independent assays.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is a key glycolytic enzyme but can also be involved in other central pathways of carbon metabolism [3].
This enzyme is responsible for the oxidative phosphorylation of G3P in the presence of NAD+ and inorganic phosphate.
In the present work, GAPDH from Idiomarina loihiensis was purified to electrophoretic homogeneity from a soluble protein fraction of approximately 240 mg corresponding to approximately 72 U GAPDH, and purification fold of 6.
The purification of the enzyme was carried out by a simple procedure involving only one chromatography step. As for other NAD+ dependent GAPDHs [10,20,22], dye-affinity chromatography on Blue Sepharose was very effective for this purification and no additional steps were required to obtain homogeneous GAPDH samples. However, a slight modification in the protocol to improve our purification is yield. Indeed, the previously cited buffer (pH 7.5) used to wash the column and the elution was performed with 10 mM NAD+ in a buffer containing: 25 mM Tris-HCl, 2 mM EDTA, 10 Mm β mercaptoethanol but at (pH 9.5). As stated above, SDSPAGE of the purified enzyme showed a single band corresponding to a 36 kDa protein [Figure 1(a), Lane 4].
This result, compared to the native molecular weight (147 kDa), suggests that the enzyme has a homotetrameric structure like other GAPDHs [9,10,21]. However, Idiomarina loihiensis enzyme sub-unit had an estimated molecular weight of approximately 36 kDa, somewhat higher than the one reported for the GAPDH subunit of different organisms like bacterial species Bacillus cereus (35 kDa) and the same weight of the amphibian Xenopus sp. (36 kDa) [23,24]. The production of the polyclonal antibodies, from the purified GAPDH Idiomarina loihiensis as immunogen, recognized a single protein band in both crude extract and purified preparations, corresponding to the GAPDH subunit. (Figure 1(b)) confirmed that the relative molecular weight of the detected protein would be the expected one for the GAPDH monomer (36 kDa).
The purified GAPDH was found to be homogeneous, suggesting the presence of one GAPDH isoform and that a single Gap C gene is expressed. Also a single GAPDH isoform has been found in some other animals tissues and microorganisms, both prokaryotes and eukaryotes [4,21,24].
However, it does not seem to be a general rule, as the presence of several GAPDH isoforms has been reported in organisms phylogenetically very different [3,4,11,2526].
The obtained value for Vmax was 2.06 U/mg. The Km of D-G3P was approximately similar to those found for cytosolic GAPDHs purified from other unicellular organisms such as Tetrahymena pyriformis [20] or mammalians like Jaculus orientalis and human tissues [12,26].
Whereas the Km of NAD+ of sardinella GAPDH is clearly higher, suggesting therefore a lower affinity for the nucleotide co-enzyme, as it was observed for pleurodeles GAPDH [21]. This indicates a possible difference in the mechanism of the catalytic reaction of sardine GAPDH with a relatively higher Vmax value compared to those of Tetrahymena, Jerboa liver, and human tissues. The kinetic parameter values obtained for the purified GAPDH differed in a number of instances from those described previously.
5. ACKNOWLEDGEMENTS
The authors would like to thank Dr. Driss Mountassif for his help in carrying out the purification-characterization of the GAPDH and also the Dr. Houda Baati for providing the Idiomarina loihiensis strain.
REFERENCES
- Forthergill-Gilmore, L.A. and Michels, P.A.M. (1993) Evolution of glycolisis. Progress in Biophysics and Molecular Biology, 59, 105-135. doi:10.1016/0079-6107(93)90001-Z
- Sirover, M.A. (2005) New nuclear functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells. Cell Biochemistry, 95, 45-52. doi:10.1002/jcb.20399
- Cerff, R. (1995) The chimeric nature of nuclear genomes and the antiquity of introns as demonstrated by GAPDH gene system. In: Go, M. and Schimmel, P., Eds., Tracing Biological Evolution in Protein and Gene Structures, Elsevier, Amsterdam,
- Marangos, P.J. and Constantinides, S.M. (1974) Multiple forms of flounder muscle glyceraldehyde-3-phosphate dehydrogenase. Subunit composition, properties, and tissue distribution of the forms. Biological Chemistry, 249, 951-958.
- Wolny, M. (1977) Effect of borate on the catalytic activities of muscle glyceraldehyde 3-phosphate dehydrogenase. European Journal of Biochemistry, 80, 551-556. doi:10.1111/j.1432-1033.1977.tb11911.x
- Allison, W.S. and Kaplan, N.O. (1964) The comparative enzymology of triosephosphate dehydrogenase. Biological Chemistry, 239, 2140-2152.
- Harris, I. (1964) Structure and catalytic activity of alcohol dehydrogenases. Nature, 203, 30-34. doi:10.1038/203030a0
- Perham, R.N. (1969) The comparative structure of mammalian glyceraldehyde-3-phosphate dehydrogenases. Biochemical Journal, 111, 17-21.
- Iddar, A., Valverde, F., Serrano, A. and Soukri, A. (2002) Expression, purification, and characterization of recombinant nonphosphorylating NADP-dependent glyceraldehyde-3-phosphate dehydrogenase from Clostridium acetobutylicum. Protein Expression and Purification, 25, 519-526.
- Soukri, A., Valverde, F., Hafid, N., Elkebbaj, M.S. and Serrano, A. (1995) Characterization of muscle glyceraldehyde-3-phosphate dehydrogenase isoforms from euthermic and induced hibernating Jaculus orientalis. Biochimica et Biophysica Acta, 1243, 161-168. doi:10.1016/0304-4165(94)00137-M
- Soukri, A., Hafid, N., Valverde, F., Elkebbaj, M.S. and Serrano, A. (1996) Evidence for a posttranslational covalent modification of liver glyceraldehyde-3-phosphate dehydrogenase in hibernating jerboa (Jaculus orientalis). Biochimica et Biophysica Acta, 1292, 177-187. doi:10.1016/0167-4838(95)00200-6
- Heinz, F. and Freimuller, B. (1982) Glyceraldehyde-3- phosphate dehydrogenase from human tissues. Methods in Enzymology, 89, 301-305. doi:10.1016/S0076-6879(82)89054-X
- Serrano, A., Mateos, M.I. and Losada, M. (1991) Differential regulation by trophic conditions of phosphorylating and non phosphorylating NADP(+)-dependent glyceroldehyde-3-phosphate dehydrogenases in Chlorella fusca. Biochemical and Biophysical Research Communications, 181, 1077-1083. doi:10.1016/0006-291X(91)92047-N
- Laemmli, U.K. (1970) Cleavage of structural proteins during assembly of the head of bacteriophage T4. Nature, 227, 680-685. doi:10.1038/227680a0
- Hedrick, J.L. and Smith, A.J. (1968) Size and charge isomer separation and estimation of molecular weights of proteins by disc gel electrophoresis. Archives of Biochemistry and Biophysics, 126, 155-164. doi:10.1016/0003-9861(68)90569-9
- Vaitukaitis, J.L. (1981) Production of antisera with small doses of immunogen: Multiple intradermal injections. Methods in Enzymology, 73, 46-52. doi:10.1016/0076-6879(81)73055-6
- Cleland, W.W. (1963) The kinetics of enzyme catalysed reaction with two or more substrates or products, nomenclature and rate equations. Biochimica et Biophysica Acta, 67, 104-137. doi:10.1016/0926-6569(63)90211-6
- Bradford, M.M. (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248-254. doi:10.1016/0003-2697(76)90527-3
- Mountassif, D., Baibai, T., Fourrat, L., Moutaouakkil, A., Iddar, A., Soukri, A. and El Kebbaj, M.S. (2009) Immunoaffinity purification and characterization of glyceraldehyde-3-phosphate dehydrogenase from human erythrocytes. Acta Biochimica et Biophysica Sinica, 41, 399- 406.
- Hafid, N., Valverde, F., Villalobo, E., Elkebbaj, M., Torres, A., Soukri, A. and Serrano, A. (1998) Glyceraldehyde-3-phosphate dehydrogenase from Tetrahymena pyriformis: Enzyme purification and characterization of a gap C gene with primitive eukaryotic features. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology, 119, 493-503.
- Mounaji, K., Erraiss, N., Iddar, A., Wegnez, M., Serrano, A. and Soukri, A. (2002) Glyceraldehyde-3-phosphate dehydrogenase from the newt Pleurodeles waltl. Protein purification and characterization of a GapC gene. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology, 131, 411-421. doi:10.1016/S1096-4959(01)00518-8
- Thompson, S.T., Cass, K.H. and Stellwagen, E. (1975) Blue dextran-sepharose: An affinity column for the dinucleotide fold in proteins. Proceedings of the National Academy of Sciences, 72, 669-672. doi:10.1073/pnas.72.2.669
- Nickells, R.W., Browder, L.W. and Wang, T.I. (1989) Factors influencing the heat shock response of Xenopus laevis embryos. Biochemistry and Cell Biology, 67, 687- 695. doi:10.1139/o89-103
- Iddar, A., Serrano, A. and Soukri, A. (2002) Phosphate-stimulated NAD(P)+-dependent glyceraldehyde-3-phosphate dehydrogenase in Bacillus cereus. FEMS Microbiology Letters, 211, 29-35. doi:10.1111/j.1574-6968.2002.tb11199.x
- Fourrat, L., Iddar, A. and Soukri, A. (2007) Purification and characterization of cytosolic glyceraldehyde-3-phosphate dehydrogenase from the dromedary camel. Acta Biochimica et Biophysica Sinica, 39, 148-154. doi:10.1111/j.1745-7270.2007.00256.x
- Serrano, A., Mateos, M.I. and Losada, M. (1993) ATPdriven transhydrogenation and ionization of water in a reconstituted glyceraldehyde-3-phosphate dehydrogenase (phosphorylating and non-phosphorylating) model system. Biochemical and Biophysical Research Communications, 197, 1348-1356. doi:10.1006/bbrc.1993.2625
NOTES
*Corresponding author.