Open Journal of Respiratory Diseases
Vol.4 No.1(2014), Article ID:43045,6 pages DOI:10.4236/ojrd.2014.41002
Impact of Cystic versus Cylindrical Types of Bronchiectasis on Pulmonary and Cardiac Functions
1Chest Department, Assiut University Hospital, Assiut, Egypt
2Internal Medicine Department, Assiut University Hospital, Assiut, Egypt
3Radiology Department, Assiut University Hospital, Assiut, Egypt
Email: *wafaagadallah@yahoo.com
Copyright (c) 2014 Wafaa Ali Hassan et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights (c) 2014 are reserved for SCIRP and the owner of the intel-lectual property Wafaa Ali Hassan et al. All Copyright (c) 2014 are guarded by law and by SCIRP as a guardian.
Received December 15, 2013; revised January 15, 2014; accepted January 22, 2014
Keywords:High Resolution Computerized Tomography; Pulmonary Function Tests; Right Ventricular Diameter; Systolic Pulmonary Artery Pressure
ABSTRACT
Background: The combination of the clinical features, HRCT score and echocardiographic evidence of pulmo-nary hypertension help assess the prognosis in bronchiectasis. Aim: To test whether pulmonary and cardiac functions associated with bronchiectasis would differ according to its type (cystic versus cylindrical) utilizing HRCT score, PFTs and echocardiography. Patients and Methods: A cross-sectional study of patients with bron-chiectasis was conducted at Chest and Internal Medicine Departments at Assiut University Hospital, Egypt. The diagnosis of bronchiectasis type was based on HRCT findings. PFTs, HRCT score and echocardiography were assessed in all cases. Results: We studied 56 patients with bronchiectasis; 31 were cystic (group A) and 25 were cylindrical (group B). Forced vital capacity (FVC%) and Dlco% were significantly lower in group A (P < 0.0001) as compared with group B; whereas FEF 25% - 75% and FEV1 (0.04) were lower in group B. Global HRCT score, RVD and SPAP were significantly higher in group A (P = 0.002) and correlated with FEV1% (r = -0.51), and with SPAP (r = 0.16). Conclusions: Airway obstruction and small airway dysfunction were more significantly seen in cylindrical bronchiectasis. Patients with cystic bronchiectasis had significantly, higher global HRCT scores, RVD and SPAP. HRCT scores correlate with FEV 1% and SPAP and could be a predictor of future PH. Routine echocardiographic assessment of patients with bronchiectasis, particularly in those with cystic disease is highly recommended.
1. Introduction
High-resolution CT (HRCT) of the chest has become the imaging technique of choice, in the diagnosis of bronchiectasis. It helps to detect findings that are not seen on plain chest radiographs, as well as classify different pathological types and determine the extent of bronchiectasis [1,2]. The combination of the clinical features of severe lung disease and echocardiographic evidence of pulmonary hypertension or right ventricular dysfunction is associated with a poor prognosis. Several different echocardiographic techniques have been used to assess right ventricular function and pulmonary artery pressure in bronchiectasis. Doppler echocardiography estimates of pulmonary artery pressure using the peak velocity of tricuspid regurgitant jet are a reliable non-invasive way of measuring pulmonary artery pressure [3]. Due to disturbed pulmonary physiology, these patients may show airflow obstruction or mixed obstructive and restrictive defect [4]. Differentiation between cystic and cylindrical bronchiectasis using HRCT score, PFTs and echocardiography was not fully discussed. We therefore carried out this prospective cross-sectional study to test whether pulmonary and cardiac functions associated with bronchiectasis would differ according to its type (cystic versus cylindrical) utilizing HRCT score, PFTs and echocardiography.
2. Patients and Methods
This prospective cross-sectional study included 56 patients (37 men and 19 women) with bronchiectasis. All patients were admitted to chest and internal medicine departments of Assiut University Hospitals, from May 2011 to November 2012. All patients were subjected to thorough history taking and clinical chest examination. Diagnosis of bronchiectasis was based upon clinical, CXR, and HRCT findings, all cases had the same degree of the number of affected lobes (≥2 lobes). According to HRCT, patients were classified as either having cystic bronchiectasis (group A) or having cylindrical bronchiectasis (group B). HRCT, echocardiography, and pulmonary function tests were done to all cases.
2.1. Pulmonary Function Tests
Forced Spirometry and the single-breath Carbon monoxide diffusing capacity of the lung (DLco) were obtained with pulmonary function units (Spirolab MIR, Italy). The Forced vital capacity (FVC) and DLCO were expressed as a percentage of predicted value based on height, age, gender and ethnic origin. Measurement of forced vital capacity (FVC) and forced vital capacity in 1 second (FEV1; best results of 3 successful attempts) was done in all patients. Total lung capacity (TLC) and residual volume (RV) were measured by plethysmography to diagnose cases with mixed obstructive and restrictive defects. Data were expressed as percentage of predicted value using the standard protocol of the American Thoracic Society [5]. Arterial blood gases were measured while patients were breathing room air. Hypoxemia and hypercarpia were defined as PaO2 < 60 mm Hg and PaCO2 > 45 mm Hg respectively.
2.2. HRCT Interpretation
HRCT scans were obtained on 16 rows HRCT (GE medical systems) scanner. Images were obtained at 1 mm collimation at 1-mm intervals from the apices to the lung bases, 0.5-second gantry rotation time, 120 kVp, and 130 mAs during maximum inspiration with the patient supine and extending from lung apex to diaphragm. All images were displayed at a window level of –700 Hounsfield units (HU) and at window width of 1500 HU, as appropriate for lung parenchyma. Two radiologists who were blinded to the clinical and laboratory data, used according to recommendations of the Nomenclature Committee of the Fleischner Society [1], The CT scan was interpreted for the presence of bronchiectasis severity, pattern, distribution, and associated disease processes such as emphysema and small airway disease. Bronchiectasis was defined as a bronchus with an internal diameter larger than its accompanying pulmonary artery, lack of tapering of the bronchial lumen for longer than 2 cm, and visualization of a bronchus within 1 cm of the costal pleura. Peribronchial thickening was characterized by a bronchial wall thicker than 1 mm. Cylindrical bronchiectasis was diagnosed based on dilatation and thickening of the bronchial wall arterial wall ratio > 1 [6]; and cystic bronchiectasis was diagnosed by noticing thin-walled cystic spaces that may contain fluid and these were seen in subsequent axial cuts either in a conglomerate fashion or in branching order [7].
The HRCT scoring system used in the present study was a modified Bhalla CT scoring system [8] Six CT scan criteria were assessed: 1) bronchial dilatation, 2) peribronchial wall thickening, 3) number of bronchiectasis segments, 4) number of bullae, 5) number of emphysema segments, and 6) criteria of associated small airway disease. Each of the above parameters were scored from 0 in absence of a lesion through 3 according to severity of disease, with the small airway disease scored as 0 if it was absent or 1 if it was present. A global score for each patient was calculated from these measures that reflected the radiological overall severity of the disease process and its associations, with a maximum possible score of 16 [8].
2.3. Echocardiography
Two-dimensional transthoracic cardiographs with color flow imaging were performed in all patients (Philips model 5500). The ECHO study was read by a cardiologist without information about patient status. Pulmonary hypertension (PH) was defined in this study as SPAP ≥ 40 mm Hg based on criteria established by the World Health Organization Symposium on Primary Pulmonary Hypertension [9]. SPAP was calculated based on the modified Bernoulli equation, and right atrial pressure was estimated as 5, 10, 15, or 20 mm Hg on the basis of size and respiratory changes of the inferior vena cava using previously described techniques [3].
3. Results
This study included 56 patients, of whom 37 (68%) were men and 19 (32%).were women. The mean age (±SD) was (53.6 ± 13.9) years. Cystic bronchiectasis (Group A, Figure 1) was diagnosed in 31 (55%) and cylindrical bronchiectasis (Group B, Figure 2) in 25 (45%) patients. No significant difference was detected between both groups regarding mean age, sex, smoking history or disease extension (numbers of affected lobes). Pulmonary function tests data of all patients are shown in Table 1. Obstructive pulmonary dysfunction was seen in a major-
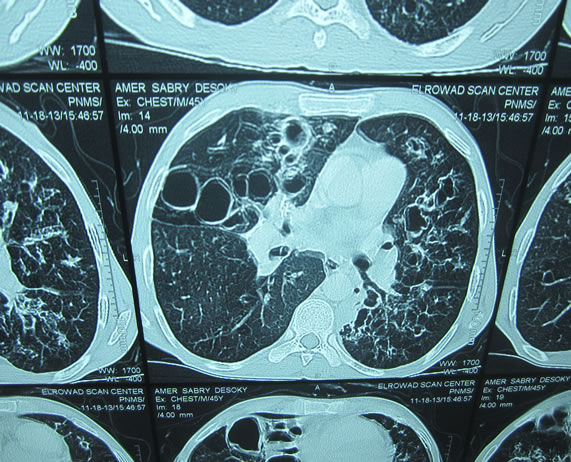
Figure 1. Cystic bronchiectasis.

Figure 2. Cylindrical bronchiectasis.
ity of patients (n = 28, 50%), whereas 20% has restrictive defects (n = 12), mixed obstructive restrictive dysfunction in 18% (n = 10) and pulmonary functions was normal in 6 cases (12%). There was a statistically significant reduced mean values in group B in relation to FEV1%, FEV1/FVC and FEF 25% - 75% (0.000). However, DLCO% and FVC% mean values were more significantly reduced in group A (0.03). There was a statistically significant increase in mean PASP, RVD and mean global HRCT score in group A (Table 2). Global HRCT scores were statistically significantly negatively correlated with the values of FEV1% (r = −0.52, p = 0.03) and significantly positively correlated with SPAP in group A (r = 0.16, p = P = 0.000). Right ventricular (RV) systolic dysfunction was observed in 21 patients in group A (67%) and in 7 patients of group B (28%) whereas left ventricular (LV) systolic dysfunction was observed in 4 patients (12%), and LV diastolic dysfunction was observed in 5 patients (16%); all had cystic bronchiectasis (group
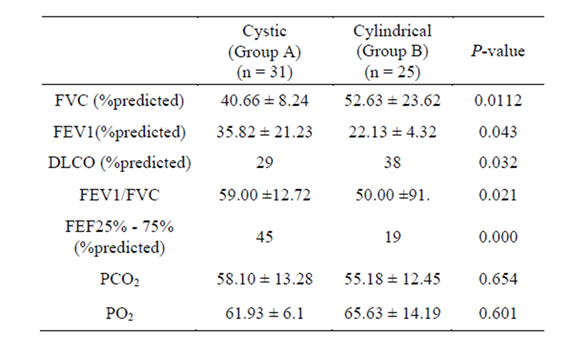
Table 1. Pulmonary function tests data in cystic and cylindrical bronchiectasis.
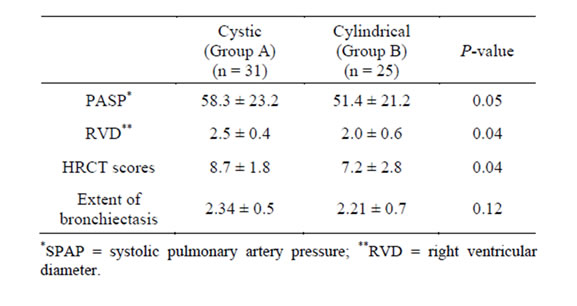
Table 2. PASP, RVD and HRCT score in cystic and cylindrical bronchiectasis.
A). In group A (cystic bronchiectasis), RVD was positively correlated with SPAP (r = 0.63, p = 0.000) and negatively correlated with PaO2 (r = −0.47, p = 0.004). In cystic bronchiectasis, SPAP was positively correlated with PaCO2 (r = 0.81, p = 0.000), and inversely correlated with PaO2 (r = −610, p = 0.000), and FEV1 (r = −721, p = 0.001).
4. Discussion
HRCT scans directly quantify lung structural abnormalities associated with bronchiectasis and PFTs indirectly measure pulmonary function to give a global assessment and can be even normal in milder disease [10]. In this study, obstructive pulmonary defect was reported in a majority of patients with bronchiectasis (50%), and FEV1%, FVC/FEV1 and FEF 25% - 75% were significantly lower in patients with cylindrical bronchiectasis as compared with the cystic group. Spirometry results were normal in 6 patients while HRCT scans showed bronchiectasis. Lynch et al. showed that airflow obstruction was common in a majority of his study group (33 patients) [11]. The cause of airflow obstruction, most likely bronchiolitis, secretion, and bronchial hyper-responsiveness [12,13]. Hansel et al. [14] studied areas of decreased attenuation using HRCT and concluded that small airway disease and inflammatory bronchiolitis are integral part of bronchiectasis. In their study, bronchiolitis was also found in lobes that were free of bronchiectasis, suggesting that bronchiolitis may precede the development of bronchiectasis and that small airways are affected before large airways. Small airway dysfunction was much observed with cylindrical bronchiectasis in our study (p = 0.000). The cystic group had lower DLCO% and FVC% than the cylindrical group. These further support the restrictive component of their disease, which is presumably related to fibrosis, representing a late irreversible disease commonly seen in these patients [15].
Hypoxemia and hypercapnia were more seen in cystic group. A previous study of regional ventilation in bronchiectatic patients using Xenon-133 scintigraphy demonstrated reduced ventilation that was widespread even in nonbronchiectasis areas [16].
Ashore studied hemodynamics in bronchiectasis using V/Q scan and pulmonary angiography [17]. He found that the cystic group had more capillary bed destruction and extensive anastomosis between pulmonary and bronchial arteries, with greater likelihood of compromised gas exchange [18]. Furthermore, in advanced bronchiectasis the respiratory muscles are exposed repetitively to increased work because of hyperinflation and alteration of the length-tension relationship [19].
In this study, patients with cystic disease were found to have significantly higher HRCT scores. We also found a negative correlation between HRCT score and FEV1%. This is in agreement with previous reports; as the HRCT score reflects bronchiolitis, areas of emphysema, bullae, and bronchiectatic segments [20]—all can affect FEV1% measurements [21].
In this study, SPAP was positively correlated with HRCT score in cystic group. PH is commonly seen in advanced bronchiectasis, particularly with cystic disease [22]. It was reported that 32.9% of stable patients with bronchiectasis had PH and SPAP was higher in cystic bronchiectasis with concomitant right and left ventricular dysfunction [23]. The pathogenesis of PH in these patients is related to impairment of pulmonary physiology due to extensive lung damage and high pulmonary vascular resistance. This is presumably due to extensive anastomosis between pulmonary and bronchial arteries and the presence of hypoxemia, which further contribute to higher SPAP [24,25].
In this study, RV systolic dysfunction and PH were more common than LV dysfunction in patients with cystic bronchiectasis. RV systolic function has not been systematically investigated in those patients. Tomlin et al. [26] described chronic cor pulmonale as a complication of fibrocystic disease of the pancreas in 1952. Vizza et al. [27] examined RV and LV function in patients with severe airway, parenchymal, and pulmonary vessel disease; the presence of RV dysfunction varied between 59% and 94%. Koelling et al. [28] observed that RV systolic function at rest and after peak exercise was similarly compromised in 40 patients with advanced cystic fibrosis, as well as in 9 patients with moderately severe bronchiectasis. They also noticed abnormal diastolic function in patients with cystic fibrosis compared with control subjects or patients with bronchiectasis. In their study, however, patients with cystic fibrosis had greater impairment of pulmonary function than those with bronchiectasis.
In cystic group, a close correlation between RV dimensions and both PaO2, and PASP These findings confirms the important role of hypoxemia and PH in the pathophysiology of RV dysfunction, which has been described before in other pulmonary diseases [23].
Previous studies in patients with other chronic lung diseases regarded LV dysfunction are rare. Vizza et al. [27] reported that LV dysfunction was present in 6% of patients with advanced stage of different pulmonary diseases, with a higher prevalence in patients with PH.
In this study, 4 patients had LV systolic dysfunction and 5 patients had LV diastolic dysfunction. Nevertheless, these findings suggest that abnormal LV performance may be due to ventricular interdependence, in which RV dilatation leads to bulging of the septum into the LV, which in turn increases LVEDD, decreases LV performance, and alters left atrium mechanics. This is in agreement with previous reports [28,29] on the cause of LV diastolic dysfunction in patients with PH.
There was negative correlation of SPAP with FEV1. This negative correlation was seen in cystic disease, whereas no correlation was found in cylindrical bronchiectasis, suggesting that the latter disease has a more favorable course.
SPAP correlated negatively with FEV1 in patients with cystic bronchiectasis. A previous study [30] on bronchiectasis emphasized that obstructive pulmonary insufficiency was related to morphologic changes and bronchial responsiveness. Obliterative bronchiolitis of small and medium airways together with secretions and associated emphysematous changes all contribute to pulmonary function deterioration associated with obstructive disease [31,32]. The above findings of deteriorating pulmonary physiology, particularly in patients with cystic bronchiectasis, have an important role in the pathogenesis of PH and suggest that PH is a marker of lung damage in these patients.
5. Conclusion
In conclusion, cystic bronchiectasis is associated with lung function impairment and worse HRCT scores as compared with cylindrical bronchiectasis. HRCT scores correlate with FEV1% and SPAP and could be a predictor of future PH. PH, therefore, can be a marker of lung damage in bronchiectatic patients. Detailed echocardiographic assessment of patients with bronchiectasis, particularly in those with cystic disease is recommended. This cross-sectional study does not allow us to determine the outcome in our patients, particularly in those who had PH. Future studies may focus on this point. Lack of follow-up of patients is a limitation of this study. Larger sample size, multicentric studies are needed to confirm our results.
REFERENCES
- A. B. Chang, J. P. Masel, N. C. Boyce, et al., “Non-CF Bronchiectasis Clinical and HRCT Evaluation,” Pediatric Pulmonology, Vol. 35, No. 6, 2003, pp. 477-483. http://dx.doi.org/10.1002/ppul.10289
- J. S. Kim, N. L. Müller, C. S. Park, P. Grenier and C. J. Herold, “Cylindrical Bronchiectasis: Diagnostic Findings on Thin-Section CT,” American Journal of Roentgenology, Vol. 168, No. 3, 1997, pp. 751-754. http://dx.doi.org/10.2214/ajr.168.3.9057529
- A. A. Ionescu, N. Payne, I. Obieta-Fresnedo, et al., “Subclinical Right Ventricular Dysfunction in Cystic Fibrosis. A Study Using Tissue Doppler Echocardiography,” American Journal of Respiratory and Critical Care Medicine, Vol. 163, No. 5, 2001, pp. 1212-1218. http://dx.doi.org/10.1164/ajrccm.163.5.9908005
- H. R. Roberts, A. U. Wells, D. G. Milne, M. B. Rubens, J. Kolbe, P. J. Cole, et al., “Air Flow Obstruction in Bronchiectasis: Correlation between Computed Tomography Features and Pulmonary Function Tests,” Thorax, Vol. 55, 2000, pp. 198-204. http://dx.doi.org/10.1136/thorax.55.3.198
- R. Pellegrion, G. Viegi, P. Enright, et al., “Interpretation of Lung Function Test,” European Respiratory Journal, Vol. 26, No. 5, 2005, pp. 948-968. http://dx.doi.org/10.1183/09031936.05.00035205
- M. Bhalla, N. Turcios, V. Aponte, et al., “Cystic Fibrosis: Scoring System with Thin-Section CT,” Radiology, Vol. 179, 1991, pp. 783-788.
- L. Marti-Bonmati, F. J. Catala and F. Ruiz Perales, “Computed Tomography Differentiation between Cystic Bronchiectasis and Bullae,” Journal of Thoracic Imaging, Vol. 7, 1991, pp. 83-85. http://dx.doi.org/10.1097/00005382-199112000-00011
- J. H. Austin, N. L. Muller, P. J. Friedman, et al., “Glossary of Terms for CT of the Lungs: Recommendations of the Nomenclature Committee of the Fleischner Society,” Radiology, Vol. 200, 1996, pp. 327-331.
- A. H. Alzeer, A. F. Al-Mobeirek, H. A. Al-Otair, U. A. Elzamzamy, I. A. Joherjy and A. S. Shaffi, “Right and left Ventricular Function and Pulmonary Artery Pressure in Patients with Bronchiectasis,” Chest, Vol. 133, No. 2, 2008, pp. 468-473. http://dx.doi.org/10.1378/chest.07-1639
- R. Pellegrion, G. Viegi, P. Enright, et al., “Interpretation of Lung Function Test,” European Respiratory Journal, Vol. 26, 2005, pp. 948-968.
- D. A. Lynch, J. Newell, V. Hale, D. Dyer, K. Corkery, N. L. Fox, et al., “Correlation of CT Findings with Clinical Evaluations in 261 Patients with Symptomatic Bronchiectasis,” American Journal of Roentgenology, Vol. 173, No. 1, 1999, pp. 53-58. http://dx.doi.org/10.2214/ajr.173.1.10397099
- L. I. Landau, P. D. Phelan and H. E. William, “Ventilatory Mechanics in Patients with Bronchiectasis Starting in Childhood,” Thorax, 2011, pp. 304-307.
- E. Y. Kang, R. R. Miller and N. L. Müller, “Bronchiectasis: Comparison of Preoperative Thin-Section CT and Pathologic Findings in Resected Specimens,” Radiology, Vol. 195, 2010, pp. 649-654.
- D. M. Hansell, A. U. Wells, M. B. Rubens and P. J. Cole, “Bronchiectasis: Functional Significance of Areas of Decreased Attenuation at Expiratory CT,” Radiology, Vol. 193, 1994, pp. 369-374.
- T. E. Robinson, A. N. Leung, W. H. Northway, F. G. Blankenberg, D. A. Bloch, J. W. Oehlert, et al., “Spirometer-Triggered High-Resolution Computed Tomography and Pulmonary Function Measurements during an Acute Exacerbation in Patients with Cystic Fibrosis,” The Journal of Pediatrics, Vol. 138, No. 4, 2001, pp. 553-559. http://dx.doi.org/10.1067/mpd.2001.111820
- H. Bass, J. A. Henderson, T. Heckscher, A. Oriol and N. R. Anthonisen, “Regional Structure and Function in Bronchiectasis: A Correlative Study Using Bronchography and 133Xe,” American Review Respiratory Disease, Vol. 97, 1968, pp. 598-609.
- M. Ashour, “Hemodynamic Alterations in Bronchiectasis: A Base for a New Subclassification of the Disease,” The Journal of Thoracic and Cardiovascular Surgery, Vol. 112, No. 2, 1996, pp. 328-334. http://dx.doi.org/10.1016/S0022-5223(96)70258-1
- J. Bahous, A. Cartier, L. Pineau, C. Bernard, H. Ghezzo, R. R. Martin, et al., “Pulmonary Function Tests and Airway Responsiveness to Methacholine in Chronic Bronchiectasis of the Adult,” Bull Eur Physiopathol Respir, Vol. 20, 1984, pp. 375-380.
- N. M. Braun, N. S. Arora and D. F. Rochester, “ForceLength Relationship of the Normal Human Diaphragm,” Journal of Applied Physiology, Vol. 53, 1982, pp. 405- 412.
- N. G. Koulouris, S. Retsou, E. Kosmas, K. Dimakou, K. Malagari, G. Mantzikopoulos, et al., “Tidal Expiratory Flow Limitation, Dyspnoea and Exercise Capacity in Patients with Bilateral Bronchiectasis,” European Respiratory Journal, Vol. 21, No. 5, 2003, pp. 743-748. http://dx.doi.org/10.1183/09031936.03.00301103
- P. Loubeyre, M. Paret, D. Revel, T. Wiesendanger and J. Brune, “Thin-Section CT Detection of Emphysema Associated with Bronchiectasis and Correlation with Pulmonary Function Tests,” Chest, Vol. 109, No. 2, 1996, pp. 360-365. http://dx.doi.org/10.1378/chest.109.2.360
- S. G. Haworth, “Primary and Secondary Pulmonary Hypertension in Childhood: A Clinicopathological Reappraisal,” Current Topics in Pathology, Vol. 73, 1983, pp. 91-152. http://dx.doi.org/10.1007/978-3-642-69134-8_3
- S. W. Royce, “Corpulmonale in Infancy and Early Childhood. Report on 34 Patients with Special Reference to the Occurrence of Pulmonary Heart Disease in CF of the Pancreas,” Pediatrics, Vol. 8, 1951, pp. 255-274.
- A. A. Ionescu, N. Payne, I. Obieta-Fresnedo, et al., “Subclinical Right Ventricular Dysfunction in Cystic Fibrosis. A Study Usingtissue Doppler Echocardiography,” American Journal of Respiratory and Critical Care Medicine, Vol. 163, No. 5, 2001, pp. 1212-1218. http://dx.doi.org/10.1164/ajrccm.163.5.9908005
- R. C. Stern, G. Borkat, S. S. Hirschfeld, et al., “Heart Failure in Cystic Fibrosis: Treatment and Prognosis of Corpulmonale with Failure of the Right side of the Heart,” American Journal of Diseases of Children, Vol. 134, No. 4, 1980, pp. 267-272. http://dx.doi.org/10.1001/archpedi.1980.02130150025007
- C. E. Tomlin, R. Bic Logueu and J. W. Hurst, “Chronic Corpulmonale as a Complication of Fibrocystic Disease of the Pancreas,” American Heart Journal, Vol. 44, No. 1, 1952, pp. 42-50. http://dx.doi.org/10.1016/0002-8703(52)90170-1
- C. D. Vizza, J. P. lynch, L. L. Ochoa, et al., “Right and Left Ventricular Dysfunction in Patient with Sever Pulmonary Disease,” Chest, Vol. 113, No. 3, 2009, pp. 576-5 83. http://dx.doi.org/10.1378/chest.113.3.576
- T. M. Koelling, G. W. Des, L. C. Ginns, et al., “Left Ventriculardiastolic Function in Patient with Cystic Fibrosis,” Chest, Vol. 123, No. 5, 2003, pp. 1488-1494. http://dx.doi.org/10.1378/chest.123.5.1488
- E. K. Louie, S. S. Lin, S. I. Reynertson, et al., “Pressure and Volume Loading of Right Ventricle Have Oppos Effect on Left Ventricular Ejaculation Fraction,” Circulation, Vol. 92, 1995, pp. 819-824. http://dx.doi.org/10.1161/01.CIR.92.4.819
- M. H. Gotz, O. C. Burghuber, U. Salzer-Muhar, et al., “Corpulmonale in Cystic Fibrosis,” Journal of the Royal Society of Medicine, Vol. 82, 1989, pp. 26-31.
- A. Rosenthal, C. R. Tucker, R. G. Williams, et al., “Echocardiographic Assessment of Corpulmonale in Cystic Fibrosis,” Pediatric Clinics of North America, Vol. 23, 1976, pp. 327-344.
- D. A. Raeside, A. Smith, A. Brown, et al., “Pulmonary Artery Pressure Measurement during Exercise Testing in Patients with Suspectedpulmonary Hypertension,” European Respiratory Journal, Vol. 16, No. 2, 2001, pp. 282- 287. http://dx.doi.org/10.1034/j.1399-3003.2000.16b16.x
NOTES
*Corresponding author.