Journal of Biosciences and Medicines
Vol.07 No.07(2019), Article ID:93780,14 pages
10.4236/jbm.2019.77010
E-Cigarette Vapor Decreases Cellular Proliferation through Nicotine-Dependent Mechanisms
Stefanie Rigg, Lindsay M. Gielda*
Purdue University, Northwest, Westville, IN, USA
Copyright © 2019 by author(s) and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: June 10, 2019; Accepted: July 19, 2019; Published: July 22, 2019
ABSTRACT
Electronic cigarette (e-cigarette) use has risen as individuals look for healthier nicotine delivery systems than conventional cigarettes, but recent studies have demonstrated variable physiological responses to e-cigarette vapor. The safety of e-cigarette vapor remains an open question. We investigated the molecular cellular response of adenocarcinoma human alveolar basal epithelial (A549) cells to flavored e-cigarette vapor. E-cigarette and cigarette vapor were extracted via a vacuum-percolating system into PBS. The effects of a flavored e-liquid containing nicotine (eCSE) and nicotine free e-liquid (NFeCSE) were compared to cigarette smoke extract (CSE), pure nicotine, glycerol, and PBS. Cells exposed to eCSE and NFeCSE had decreased cell viability and proliferation. An increase in active caspase-3 was observed in cells exposed to NFeCSE, while cells exposed to eCSE and CSE displayed no change, suggesting nicotine-mediated protection from apoptosis. However, phosphorylated Akt, which regulates cellular proliferation, was elevated in response to eCSE, NFeCSE, CSE, and glycerol. As the glutathione-oxidative stress response was known to integrate cellular factors including carcinogenesis, inflammation, and cell proliferation, GSH/GSSG levels were measured in cells exposed to e-cigarette smoke. An increase in oxidative stress was observed in cells treated with CSE, but not eCSE, suggesting components in CSE independent of nicotine may be contributing to an imbalance in the stress response. These results indicate e-cigarette vapor affects A549 cell viability dependent on nicotine, and is distinct from CSE-induced mechanisms of oxidative-stress.
Keywords:
E-Cigarettes, Oxidative Stress, Nicotine, Apoptosis

1. Introduction
One of the leading causes of preventable morbidity and mortality in the U.S. is tobacco use, with the most common form being cigarette smoking. Smoking is a causative agent of multiple chronic diseases including: stroke, heart disease, pneumonia, diabetes, vascular disease, and lung disease including chronic obstructive pulmonary disease (COPD), asthma, and a variety of cancers [1] - [16] . E-cigarettes are promoted as a safer alternative to tobacco cigarettes, purportedly secondary to the production of fewer toxins and carcinogens [17] [18] . With the sudden rise in e-cigarette use, studies aimed at identifying that the risks of e-cigarette use have found mixed results.
E-cigarettes vaporize a solution, referred to as e-liquid, which contains variable components including nicotine and flavoring additives in solvents such as propylene glycol and glycerol. The composition of e-liquid, as well as concentration of nicotine in the solution, varies extensively across manufacturers. Studies investigating the effects of e-cigarettes have shown variable outcomes, dependent on the model, e-liquid composition, and delivery method. However, several studies have found that e-cigarette vapor exposure altered the redox physiology of lung epithelial cells, induced proinflammatory cytokine secretion in bronchial alveolar lavage fluid in mice, and induced gene expression in a similar manner to carcinogens [19] [20] [21] [22] .
Cigarette smoke can induce an upregulation of pro-inflammatory cytokines, as well as activation of apoptotic and necrotic signaling pathways in mammalian cell cultures in response to oxidative stress [23] [24] [25] . Prolonged oxidative stress due to cigarette smoke results in the activation of caspase-mediated apoptotic pathways [24] . However, nicotine suppresses caspase-mediated apoptosis through the upregulation of the PI3K-Akt pathway, a signal transduction cascade that promotes cellular survival and growth [23] [24] [25] . As demonstrated in A549 cells, in the presence of apoptosis-inducing oxidative damage due to cigarette smoke, nicotine up-regulation of Akt results in the phosphorylation of downstream substrates that ultimately increase cell survival and tumorigenesis [26] - [31] .
Nicotine vaporization through e-cigarettes has the potential to induce similar cellular effects seen with cigarette smoking, including the upregulation of oxidative stress responsive genes, nicotine-mediated inhibition of apoptosis and upregulation of Akt cell signaling pathways [27] [28] [29] [30] [31] . The objective of this study was to determine the effects of e-cigarette vapor exposure on A549 cells compared to cigarette smoke in terms of cell proliferation and activation of Akt and apoptotic pathways. Smoke extracted from flavored e-liquid containing nicotine (eCSE), flavored nicotine free e-liquid (NFeCSE), and cigarette smoke (CSE) were added to cultured A549 cells. Nicotine, glycerol, and PBS were used as controls to assess the contribution of the individual components of e-liquid. Changes in cellular viability and characterization of molecular pathways demonstrated e-cigarette components which decreased A549 proliferation through mechanisms dependent on nicotine and flavoring components.
2. Materials Methods
Cell Culture
A549 cells were cultured in F12K media (ATCC) to contain 10% FBS (ATCC) and 5% Penicillin and Streptomycin (ATCC). Cells were incubated in a 50 mL flask containing 20 mL media with 5% CO2 at 37˚C for 48 hours. Media was then replaced, and the cells were incubated an additional 24 - 48 hr. with 5% CO2 at 37˚C, until a confluency of 80% - 90% was reached. The media was removed, and the cells were released using 2 mL trypsin. Cells were then passaged at 20% volume in a new 50 mL flask as well as seeded in 24 or 96 well plates at 3 × 105 or 1 × 105 cells per well respectively. Plates were incubated with 5% CO2 at 37˚C for 48 hr. The media was replaced along with the addition of the test reagents and incubated an additional 48 - 72 hr.
Smoke Extraction
Cigarette smoke extract (CSE) was extracted through a modified water percolator system using vacuum filtration. The cigarette smoke was drawn through a Pasteur pipette and bubbled into 1x PBS. 25 mL of PBS was used per 1 cigarette (Marlboro Gold, 0.8 mg nicotine, Phillip Morris, Richmond, VA). Electronic cigarette smoke extract (eCSE) and nicotine free electronic cigarette smoke extract (NFeCSE) were extracted through a modified water percolator system using vacuum filtration. The vapor was drawn through a Pasteur pipette and bubbled into 1x PBS. 25 mL of PBS was used per 6 drops of e-liquid (Mitten Vapors, Titty Sprinkles 6 mg nicotine or Titty Sprinkles 0 mg nicotine). 6 mg nicotine e-liquid was used as an equivalent to 1 light type cigarette and 6 drops of e-liquid was determined to be the equivalent of smoking 1 cigarette. The CSE, eCSE and NFeCSE were stored at 4˚C.
High Performance Liquid Chromatography Analysis
Nicotine concentrations in CSE and eCSE were quantified by reverse-phase high performance liquid chromatography (HPLC). The e-juice standard containing 6 mg nicotine per 25 mL solution was diluted to a nicotine concentration of 0.6 μg/mL and 0.06 μg/mL and compared to a pure nicotine standard. HPLC and MS-MS analysis were performed by Dr. Bruce Cooper at Bindley Bioscience Center, Purdue University, West Lafayette Indiana.
Cell Viability Assay
To test cell viability, A549 cells were seeded in a Falcon 24 well plate at 3 × 105 cells/well and incubated overnight in 5% CO2 at 37˚C. Test reagent (50 μL either PBS, CSE, eCSE and NFeCSE) were added to wells, and cells were incubated in 5% CO2 at 37˚C for 48 hrs. Cells were washed with PBS 2x, trypsinized as previously described, and quantified using a hemocytometer.
Cell Proliferation Assay
Cellular proliferation was measured via Bromodeoxyurdine (BrdU) colorimetric assay kit (ExAlpha Biologicals Inc). BrdU colorimetric assay was performed in accordance with the manufacturer’s instructions. Briefly, A549 cells cultured in 96 well plates seeded at 1 × 105 cells per well for 24 hours were exposed to 1 μl of each test reagent, and 10 μl/well of the BRdU labelling solutions was added to each well. Cells are incubated for 24 hours under standard conditions. The media was aspirated from each well, and 200 μl of fixative solution is added to each well, and incubated for 30 minutes. Cells were washed 3X with 1X Tris/Tween solution, and 100 μl of anti-BrdU antibody is incubated at 1 hr at room temperature. Cells were washed 3X with 1X Tris/Tween solution. 100 μl of Peroxidase conjugated goat anti-mouse IgG was added to each well and incubated for 30 minutes at room temperature. Cells were washed 3X with Tris/Tween solution, and a final distilled water wash. 100 μl of a TMB development solution was added to each well and incubated at room temperature for 30 minutes in the dark. 100 μl of ELISA stop solution is added to each well, and incorporation of BrdU was analyzed using ThermoFisher Multiskan spectrophotometer.
Apoptosis Assay
Apoptosis was measured using the Caspase-3 colorimetric assay kit (Biovision Incorporated). Caspase-3 colorimetric assay was performed in accordance with the manufacturer’s instructions. A549 cells were seeded at 4 × 105 cells/well in 24 well plates as described previously and 50 μl of test reagents were added, and incubated for 24 hours. Trypsinized cells were quantified, resuspended in 50 μl of cell lysis buffer, and incubated for 10 minutes. Cells were centrifuged for 1 minute at 10,000 ×g. The supernatant was transferred to a new tube and protein content quantified. Each sample was diluted in cell lysis buffer to 100 μg protein in 50 μl total. 50 μl of 2X reaction buffer, and 5 μl of 4 mM DEVD-pNA substrate were added and incubated at 3˚C for 1 hour. Samples were read at 405 nm in a ThermoFisher Multiskan spectrophotometer.
Akt Assay
Active phosphorylated Akt was quantified using the Pierce Akt colorimetric in-cell ELISA kit (Thermo Scientific). ELISA was performed in accordance with the manufacturer’s instructions. Briefly, A549 cells were seeded in 96 well plates at 1 × 105 cells/well as previously described and incubated for 24 hours with 1 μl of each test reagent. Media was removed, 100 μl of 4% formaldehyde added to each well, and incubated at room temperature for 15 minutes. Wells were washed 2X with 100 μl of 1X TBS. 100 μl of 1X Permeabilization Buffer was added and incubated for 15 minutes at room temperature. Wells were washed with 100 μl of 1X TBS, and 100 μl of Quenching solution added. Following 20 minutes incubation at room temperature, cells were washed with 100 μl 1X TBS, and 100 μl of Blocking Buffer was added and incubated at room temperature for 30 minutes. The blocking Buffer was removed, and 50 μl of primary antibody were added and incubated overnight at 4˚C. Cells were washed with 100 μl of buffer, and 100 μl of Diluted HRP Conjugate was incubated for 30 minutes at room temperature. Wells were washed 3X with 200 μl of wash buffer, and 100 μl of TM substrate was added to each well. Following 10 minutes of incubation, 100 μl of Stop Solution was added, and absorbance was measured at 450 nm in a Thermo Fisher Multiskan spectrophotometer.
GSH/GSSG Assay
Glutathione (GSSG/GSH) detection kit (Enzo) was used to quantify the total GSH and GSSG in the cells when exposed to test reagents. GSSG/GSH assay was performed in accordance with the manufacturer’s instructions. Briefly, A549 cells were seeded at 3 × 105 cells/well and 50 μl of each test reagent was added to individual wells and incubated for 24 hours. Trypsinized cells were centrifuged at 300 ×g for 10 minutes, were quantified using a hemocytometer, and washed with cold PBS. Pelleted cells were suspended in 500 μl of 5% (w/v) meta-phosphoric acid, and sonicated. The suspension was centrifuged at 120,000 ×g for 5 minutes at 4˚C, and the supernatant was transferred to a clean 1.5 mL tube. To quantify oxidized glutathione, 1 μl of 2 M 4-vinylpyridine was added to 50 μl of each prepared sample and 4 μM of GSSG as a standard. To quantify total glutathione, 50 μl of each sample was added directly to a 96 well plate. Following serial dilutions, 150 μl of Reaction Mix was added to each well and absorbance read at 504 nm at 1-minute intervals for 10 minutes. Samples were compared to prepared standards.
Statistical Analysis
Data analysis were performed using the mean and standard deviation of a population. For each experiment, each sample was tested in triplicate, and experiments were repeated three times. Comparisons in the data were made using the student’s t test. Significance were determined based on p < 0.05, **p < 0.001, ***p < 0.0001 compared to the PBS treated control unless otherwise indicated.
3. Results
eCSE and NFeCSE Decrease Cell Viability and Proliferation
To determine the effects of e-cigarette vapor on human lung cells, cigarette smoke (CSE), e-cigarette vapor (eCSE), and nicotine-free eCSE vapor (NFeCSE) were extracted through a modified water percolator system using vacuum filtration into PBS. HPLC analysis was performed on smoke extracts to quantify the retention and thus delivery of nicotine to cells. To assess cell viability upon smoke exposure, increasing concentrations of CSE, eCSE, NFeCSE, or PBS (2.5%, 5%, or 10% total volume) were added to cultured A549 cells, and incubated for 72 hr. Quantified via direct cell counts, eCSE and NFeCSE treated cells display a significant reduction in cell number compared to PBS treated cells, while treatment with CSE did not have a significant effect (Figure 1). This suggests that eCSE and NFeCSE have a negative effect on cell viability compared to CSE. HPLC analysis was performed on smoke extracts to quantify the retention and thus delivery of nicotine to cells. Addition of 5% extracted smoke solution equated to 0.009 µg/mL nicotine for CSE and 0.0165 µg/mL nicotine for eCSE, and was used for subsequent analysis.
To test whether the decrease in cell viability was due to a decrease in cellular proliferation or cellular death, BrdU incorporation was examined in CSE and eCSE treated cells as a marker of cellular proliferation. CSE treated cells displayed no significant change in BrdU incorporation compared to the buffered control, while eCSE and NFeCSE both had a significant decrease in BrdU incorporation (42% and 31% respectively) (Figure 2). These results indicate a reduction in cellular growth for cells treated with e-cigarette vapor compared to the control and CSE, and are consistent with the decreased cell number observed with the direct count assay (Figure 1).
In order to ascertain whether the decrease in cell count and proliferation were due to the specific components in the e-juice, the effects of pure nicotine and glycerol (a key component in the e-juice) on A549 cells were examined. Viability of cells treated with glycerol, had no significant change compared to the control, indicating that glycerol alone was not causing the significant reduction in cell viability in cells exposed to e-cigarette vapor. Viability of cells treated with pure nicotine, eCSE and NFeCSE were significantly reduced (Figure 3). This suggests that nicotine is contributing to the significant reduction in cell number in the
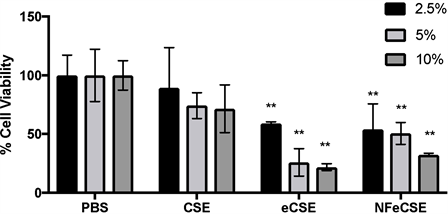
Figure 1. Effect of Vapors on Cell Viability. The effects of 5% by volume of CSE, eCSE, and NFeCSE on cell viability of A549 cells. Cells were exposed to 2.5%, 5%, or 10% total volume of the indicated extracts, incubated for 72 hr and quantified by enumeration using a hemocytometer. Data represents the percentage of cells compared to the PBS treated control, and is the mean of three independent replicates with standard deviation (n = 9). (**p < 0.001 compared to PBS treated control).
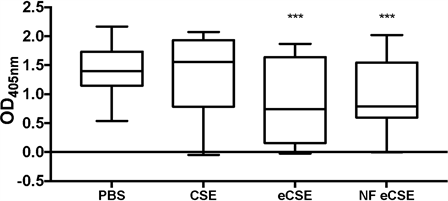
Figure 2. Effect of Vapors on Cellular Proliferation. The effects of 5% by volume of CSE, eCSE, and NFeCSE on cell proliferation of A549 cells after 24 hr, as measured by BrdU incorporation into new DNA. Data represents the range and mean OD405 with standard deviation (n = 30). (***p < 0.0001 compared to 5% by volume PBS treated control).
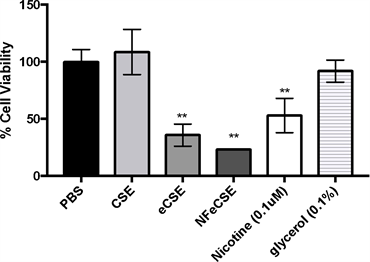
Figure 3. Effect of Individual Components of eCSE on Cellular Viability The effects of 5% by volume of CSE, eCSE, NFeCSE, 0.1 μM nicotine, and 0.1% glycerol on cell viability of A549 cells. Cells were exposed to the indicated test for 48 hr, and quantified by enumeration using a hemocytometer. Data represents the mean percentage of cells compared to the PBS treated control, and is representative of three independent replicates with standard deviation (n = 6). (**p < 0.001 compared to PBS treated control).
eCSE treated cells compared to the CSE treated cells (average values; 5% eCSE, 0.0165 µg/mL and 5% CSE, 0.009 µg/mL). Additional factors in the e-cigarette, including flavor additives, could be affecting the decrease in cell viability.
NFeCSE Increases Apoptosis in A549 Cells
Apoptosis is a tightly regulated mechanism of cell death that occurs normally during development or as a defense mechanism in response to damage. Due to the decrease in cellular proliferation and viability upon exposure to eCSE and NFeCSE, cells were examined for the activation of the apoptotic pathway. Caspase-3, the primary activator of apoptotic DNA fragmentations, is a common indicator for the activation of apoptosis. As cell count and proliferation were significantly reduced in eCSE and NFeCSE treated cells, we hypothesized that there would be an increase in active caspase-3, indicative of an increase in apoptosis. However, there was no significant change in the presence of active caspase-3 in cells exposed to eCSE and CSE compared to the control, indicating cells were not undergoing apoptosis in response to smoke exposure (Figure 4).
NFeCSE exposed cells had a significant increase (64%) in apoptosis compared to the control and eCSE. As the lack of nicotine is the only variable between NFeCSE and eCSE, the effects of nicotine on apoptosis were examined to address whether it is contributing to the observed difference. The addition of nicotine decreased levels of active caspase-3, consistent with previous reports of nicotine inhibition of caspase mediated apoptosis [25] [26] [27] . Additionally, cells exposed to glycerol had no significant change in active caspase-3. These results suggest that nicotine present in CSE and eCSE are contributing to the inhibition of caspase mediated apoptosis in A549 cells.
Akt Upregulation in Response to Glycerol
To assess the effects of e-cigarette mediated delivery of nicotine on cellular apoptosis, additional regulators of cellular survival, including activation of protein
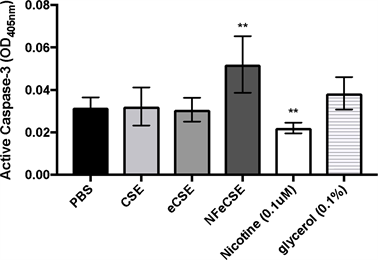
Figure 4. ENDS Effects on Apoptosis, the effects of 5% by volume of CSE, eCSE, NFeCSE, 0.1 μM nicotine, and 0.1% glycerol on active caspase-3 in A549 cells. Cells were exposed to the indicated test for 48 hrs, and active caspase-3 quantified via a colorimetric assay and reported as optical density (OD). Data represents the range and mean OD 405 nm with standard deviation (n = 6) and is representative of three independent replicates. (*p < 0.001 compared to PBS treated control).
kinase B (Akt) were examined. As Akt activity is a regulator of cellular proliferation and apoptosis, measuring phosphorylated Akt (P-Akt) in cells exposed to extracted smoke and pure components would help identify the molecular pathways responsible for the observed cell proliferation and nicotine-mediated caspase-3 inhibition. Previous studies have shown that nicotine derivatives suppress apoptosis through upregulation of the Akt pathway and inhibition of caspase-3 activation, thereby promoting cell survival [28] [29] . To test whether nicotine mediated caspase-3 inactivation seen in CSE and eCSE is due to nicotine-mediated activation of the Akt pathway, P-Akt was measured in treated cells.
Cells treated with CSE, eCSE, NFeCSE and glycerol had increased levels of P-Akt, while cells treated with nicotine had no significant change compared to PBS control (Figure 5). The results correlate with the lower levels of caspase-3 seen in the previous apoptosis assay and indicates that the decrease in cell viability observed after exposure to eCSE is due to a caspase-independent mechanism. In contrast, the decrease in cell viability after exposure to NFeCSE is due to caspase mediated apoptosis, suggesting that while lack of nicotine in NFeCSE is making cells vulnerable to apoptosis, additional components of the smoke extract can decrease cellular proliferation in A549 cells through alternative mechanisms.
Modulation of GSH:GSSG ratio in CSE Treated Cells
As cigarette smoke and nicotine have been shown to induce DNA damage due to high oxidative stress, the effects of eCSE and the role of nicotine on inducing oxidative stress in A549 cells was determined by measuring GSH and GSSG. Oxidative stress can be measured by the ratio of GSH to GSSG, where a decrease in the GSH:GSSG ratio as indicative of oxidative stress [30] . A549 cells treated with NFeCSE had an increase in GSH:GSSG compared to the PBS control, CSE, and eCSE treated (Figure 6). There was a slight decrease in GSH:GSSG in cells treated with CSE, compared to PBS and eCSE, and is consistent with other studies
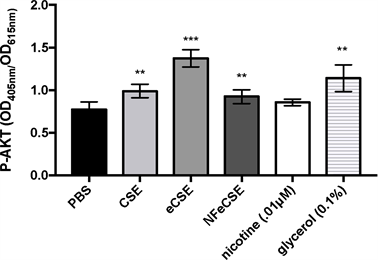
Figure 5. Akt The effects of 5% by volume of CSE, eCSE, NFeCSE, 0.1 μM nicotine, and 0.1% glycerol on P-Akt in A549 cells. Cells were exposed to the indicated test for 24 hours, and quantified by active, phosphorylated AKT(P-Akt). Data represents the range and mean OD 405/615 nm with standard deviation (n = 6) and is representative of 3 independent replicates. (**p < 0.001, ***p < 0.0001 compared to PBS treated control).
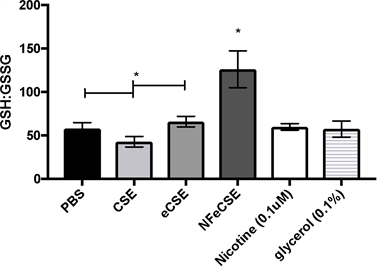
Figure 6. ENDS Effects on Oxidative Stress The effects of 5% by volume of CSE, eCSE, NFeCSE, 0.1 μM nicotine, and 0.1% glycerol on GSH and GSSG levels in A549 cells. Cells were exposed to the indicated test for 24 hr, and total GSH and GSSG quantified and reported as GSH:GSSG (pmol). Data represents the range and mean with standard deviation (n = 3) and is representative of three independent replicates. (*p < 0.05, compared to PBS, CSE, eCSE, nicotine, and glycerol.)
[31] . While these results suggest an overall decrease in oxidative stress in NFeCSE treated cells, the increase in caspase-3 activation in response to NFeCSE (Figure 4) suggests an overall imbalance of the oxidative stress response amidst apoptosis activation.
4. Discussion
E-cigarettes are perceived as a safer alternative to cigarettes and are generally believed to produce fewer harmful toxins and carcinogens in the body [17] . However, studies investigating the cellular response to e-cigarettes are limited. Since the effects of cigarette smoke and nicotine were well known, these were used as controls in examining the effects of e-cigarette vapor on A549 cells. While both eCSE and NFeCSE exposure result in a decrease in cell viability, our results suggest that this occurs through different molecular mechanisms and highlights the role of nicotine in modulating cellular pathways. NFeCSE induced caspase-3 mediated apoptosis, while this pathway was suppressed in eCSE treated cells through the upregulation of Akt [23] [24] [25] .
While no significant effects on cell viability were observed when exposed to CSE, eCSE and NFeCSE treatment resulted in a significant reduction in cell viability and proliferation. Nicotine is known to influence cell survival pathways including proliferation and apoptosis, as well as antioxidant and redox pathways in various animal models and cell lines including A549 [26] [27] [28] [32] [33] [34] . Pure liquid nicotine was used as a control, however production of nicotine derivatives due to vaporization of e-cigarettes and heating of cigarettes could result in variable results [35] . The concentrations of nicotine in the smoke and e-cigarette vapor were determined using HPLC analysis and were biologically relevant concentrations being delivered to the cells in our model [36] .
Exposure of cells to pure 0.1 μM nicotine significantly reduced cellular viability compared to PBS but could not explain the reduction in cell number of eCSE exposed cells as NFeCSE had a similar decrease in viability. The decrease in cellular proliferation of nicotine-exposed cells was surprising, as nicotine and its derivatives are known to induce cellular proliferation and inhibit caspase-mediated apoptosis [25] [26] [27] . However, a recent study has shown that nicotine had significant growth inhibitory effects on A549 cells at doses of 0.1 μM and higher [37] . In this study, Gao et al. noted apoptotic phenotypic changes of the cell including stress fiber formation and apoptotic blebs in a dose-dependent manner in cells treated with 1 μM or more. These morphological changes were not shown in cells treated with 0.1 μM nicotine. Our findings are in support of Gao et al., indicating that A549 cell exposure to 0.1 μM nicotine significantly inhibited cell proliferation and viability in a caspase-independent manner, and suggest that other components of eCSE and NFeCSE, including glycerol and flavoring agents, could be affecting cellular pathways.
Cigarette smoke and nicotine are known to affect oxidative stress pathways in several models, increasing reactive oxygen species and resulting in damage to intracellular proteins and DNA [19] [36] [38] . Glutathione neutralizes free radicals by acting as an electron donor. In a resting cell, glutathione is almost exclusively in its reduced form. In response to reactive oxygen species, antioxidative enzymes and genes involved in GSH homeostasis are upregulated, including glutathione reductase. Nicotine has been shown to inhibit glutathione reductase, which catalyzes the reduction of oxidized glutathione disulfide (GSSG) back to the reduced form, replenishing free GSH levels [39] . Therefore, in the presence of nicotine, there is a decrease in GSH:GSSH, indicative of oxidative stress. While a change in GSH:GSSG levels in cells exposed to pure nicotine was not observed, there was a slight increase in oxidative stress in cells treated with CSE, as denoted by a decrease in the GSH:GSSG ratio. NFeCSE exposed cells had a decrease in oxidative stress, as measured by an increase in the ratio of GSH:GSSG, compared to PBS, CSE, eCSE, and nicotine treated cells. These results could be an artifact of general cellular stress, as NFeCSE treated cells were shown to have an increase in caspase-3 activation (Figure 4) and decrease in cell viability (Figure 3). Alternatively, the absence of oxidative stress induction in our model could be due differences in our model compared to other studies. Our findings are in accordance with a study by Tayler et al., which found that nicotine flavored e-liquid resulted in lower oxidative stress in H292 human bronchial epithelial cells than in cells exposed to cigarette smoke [40] . Additional studies into the varying effects of flavor and solvent additives of e-liquid, on oxidative stress would aid in determining the overall effect of e-cigarettes on A549 cells.
This study found that while exposure to vapor induces a cellular response, the specific mechanism is dependent on vapor components, and highlights the limitations of this study and the necessity for ongoing research in this area. This works serves as a foundational study and current work is aimed at testing variables associated with vapor components; including the inclusion of multiple brands of cigarettes and e-cigarette liquid, and the composition of e-cigarette liquid (i.e. varying nicotine content, flavoring, solvents, and other additives). Assessment of cell viability, caspase activation, and oxidative stress of A549 cells with the inclusion of these variables will aide in determining the molecular cellular response to e-cigarette vapor.
With the results of our study indicating e-cigarettes cause an increase in cellular death, it begs the question whether e-cigarettes are a safer alternative to cigarette usage, especially in light of the effects of nicotine alone. However, the induction of cell death of a cancer-derived cell line in this study highlights the possible application of e-cigarette technology in the development of novel therapeutic cancer treatments. Further study into the effects of exposure of e-cigarettes on cancerous and non-cancerous cell lines, in addition to the effects of the variable e-liquid composition, is warranted.
Conflicts of Interest
The authors declare no conflicts of interest regarding the publication of this paper.
Cite this paper
Rigg, S. and Gielda, L.M. (2019) E-Cigarette Vapor Decreases Cellular Proliferation through Nicotine-Dependent Mechanisms. Journal of Biosciences and Medicines, 7, 121-134. https://doi.org/10.4236/jbm.2019.77010
References
- 1. Flaherty, K.R., et al. (2014) Smoking-Related Idiopathic Interstitial Pneumonia. European Respiratory Journal, 44, 594-602. https://doi.org/10.1183/09031936.00166813
- 2. Akter, S., et al. (2015) Correction: Smoking, Smoking Cessation, and the Risk of Type 2 Diabetes among Japanese Adults: Japan Epidemiology Collaboration on Occupational Health Study. PLoS ONE, 10, e0137039. https://doi.org/10.1371/journal.pone.0137039
- 3. Messner, B. and Bernhard, D. (2014) Smoking and Cardiovascular Disease: Mechanisms of Endothelial Dysfunction and Early Atherogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology, 34, 509-515. https://doi.org/10.1161/ATVBAHA.113.300156
- 4. Kwon, Y., et al. (2016) Association of Smoking, Alcohol, and Obesity with Cardiovascular Death and Ischemic Stroke in Atrial Fibrillation: The Atherosclerosis Risk in Communities (ARIC) Study and Cardiovascular Health Study (CHS). PLoS ONE, 11, e0147065. https://doi.org/10.1371/journal.pone.0147065
- 5. Rennard, S.I. and Drummond, M.B. (2015) Early Chronic Obstructive Pulmonary Disease: Definition, Assessment, and Prevention. The Lancet, 385, 1778-1788. https://doi.org/10.1016/S0140-6736(15)60647-X
- 6. Wylam, M.E., et al. (2015) Mechanisms of Cigarette Smoke Effects on Human Airway Smooth Muscle. PLoS ONE, 10, e0128778. https://doi.org/10.1371/journal.pone.0128778
- 7. Mannami, T., et al. (2004) Cigarette Smoking and Risk of Stroke and Its Subtypes among Middle-Aged Japanese Men and Women: The JPHC Study Cohort I. Stroke, 35, 1248-1253. https://doi.org/10.1161/01.STR.0000128794.30660.e8
- 8. Lee, Y.C., et al. (2009) Active and Involuntary Tobacco Smoking and Upper Aerodigestive Tract Cancer Risks in a Multicenter Case-Control Study. Cancer Epidemiology, Biomarkers & Prevention, 18, 3353-3361. https://doi.org/10.1158/1055-9965.EPI-09-0910
- 9. Hecht, S.S. (2006) Cigarette Smoking: Cancer Risks, Carcinogens, and Mechanisms. Langenbeck’s Archives of Surgery, 391, 603-613. https://doi.org/10.1007/s00423-006-0111-z
- 10. Trichopoulos, D., et al. (2011) Hepatocellular Carcinoma Risk Factors and Disease Burden in a European Cohort: A Nested Case-Control Study. Journal of the National Cancer Institute, 103, 1686-1695. https://doi.org/10.1093/jnci/djr395
- 11. Ladeiras-Lopes, R., et al. (2008) Smoking and Gastric Cancer: Systematic Review and Meta-Analysis of Cohort Studies. Cancer Causes Control, 19, 689-701. https://doi.org/10.1007/s10552-008-9132-y
- 12. Macleod, L.C., et al. (2013) Risk Factors for Renal Cell Carcinoma in the Vital Study. Journal of Urology, 190, 1657-1661. https://doi.org/10.1016/j.juro.2013.04.130
- 13. Ye, X., Lu, G., Huai, J. and Ding J. (2015) Impact of Smoking on the Risk of Pancreatitis: A Systematic Review and Meta-Analysis. PLoS ONE, 10, e0124075. https://doi.org/10.1371/journal.pone.0124075
- 14. Freedman, N.D., et al. (2011) Association between Smoking and Risk of Bladder Cancer among Men and Women. JAMA, 306, 737-745. https://doi.org/10.1001/jama.2011.1142
- 15. Oh, H.Y., et al. (2016) Association of Combined Tobacco Smoking and Oral Contraceptive Use with Cervical Intraepithelial Neoplasia 2 or 3 in Korean Women. Journal of Epidemiology, 26, 22-29.
- 16. Kondo, H., et al. (2016) Smoking Is a Risk Factor for Development of Adult T-Cell Leukemia/Lymphoma in Japanese Human T-Cell Leukemia Virus Type-1 Carriers. Cancer Causes Control, 27, 1059-1066. https://doi.org/10.1007/s10552-016-0784-8
- 17. Hajek, P., Etter, J.-F., Benowitz, N., Eissenberg, T. and McRobbie, H. (2014) Electronic Cigarettes: Review of Use, Content, Safety, Effects on Smokers and Potential for Harm and Benefit. Addiction, 109, 1801-1810. https://doi.org/10.1111/add.12659
- 18. Tomashefski, A. (2016) The Perceived Effects of Electronic Cigarettes on Health by Adult Users: A State of the Science Systematic Literature Review. Journal of the American Association of Nurse Practitioners, 28, 510-515. https://doi.org/10.1002/2327-6924.12358
- 19. Scheffler, S., et al. (2015) Evaluation of E-Cigarette Liquid Vapor and Mainstream Cigarette Smoke after Direct Exposure of Primary Human Bronchial Epithelial Cells. International Journal of Environmental Research and Public Health, 12, 3915-3925. https://doi.org/10.3390/ijerph120403915
- 20. Lerner, C.A., et al. (2015) Vapors Produced by Electronic Cigarettes and e-Juices with Flavorings Induce Toxicity, Oxidative Stress, and Inflammatory Response in Lung Epithelial Cells and in Mouse Lung. PLoS ONE, 10, e0116732. https://doi.org/10.1371/journal.pone.0116732
- 21. Wang, H., et al. (2015) Cytotoxicity of Chronic Exposure to 4 Cigarette Smoke Condensates in 2 Cell Lines. International Journal of Toxicology, 34, 182-194. https://doi.org/10.1177/1091581815574349
- 22. Garcia-Arcos, I., et al. (2016) Chronic Electronic Cigarette Exposure in Mice Induces Features of COPD in a Nicotine-Dependent Manner. Thorax, 71, 1119-1129. https://doi.org/10.1136/thoraxjnl-2015-208039
- 23. Gould, N.S., et al. (2015) Glutathione Depletion Accelerates Cigarette Smoke-Induced Inflammation and Airspace Enlargement. Toxicological Sciences, 147, 466-474. https://doi.org/10.1093/toxsci/kfv143
- 24. Ryter, S.W., et al. (2007) Mechanisms of Cell Death in Oxidative Stress. Antioxidants & Redox Signaling, 9, 49-89. https://doi.org/10.1089/ars.2007.9.49
- 25. Hakki, A., et al. (2001) Nicotine Inhibition of Apoptosis in Murine Immune Cells. Experimental Biology and Medicine (Maywood), 226, 947-953. https://doi.org/10.1177/153537020122601011
- 26. Zhang, J., Kamdar, O., Le, W., Rosen, G.D. and Upadhyay, D. (2009) Nicotine Induces Resistance to Chemotherapy by Modulating Mitochondrial Signaling in Lung Cancer. American Journal of Respiratory Cell and Molecular Biology, 40, 135-146. https://doi.org/10.1165/rcmb.2007-0277OC
- 27. Dasgupta, P., et al. (2009) Nicotine Induces Cell Proliferation, Invasion and Epithelial-Mesenchymal Transition in a Variety of Human Cancer Cell Lines. International Journal of Cancer, 124, 36-45. https://doi.org/10.1002/ijc.23894
- 28. Nakada, T., et al. (2012) Lung Tumorigenesis Promoted by Anti-Apoptotic Effects of Cotinine, a Nicotine Metabolite through Activation of PI3K/Akt Pathway. The Journal of Toxicological Sciences, 37, 555-563.
- 29. West, K.A., et al. (2003) Rapid Akt Activation by Nicotine and a Tobacco Carcinogen Modulates the Phenotype of Normal Human Airway Epithelial Cells. Journal of Clinical Investigation, 111, 81-90. https://doi.org/10.1172/JCI200316147
- 30. Zitka, O., et al. (2012) Redox Status Expressed as GSH:GSSG Ratio as a Marker for Oxidative Stress in Paediatric Tumour Patients. Oncology Letters, 4, 1247-1253. https://doi.org/10.3892/ol.2012.931
- 31. Aspera-Werz, R.H., et al. (2018) Nicotine and Cotinine Inhibit Catalase and Glutathione Reductase Activity Contributing to the Impaired Osteogenesis of SCP-1 Cells Exposed to Cigarette Smoke. Oxidative Medicine and Cellular Longevity, 2018, Article ID: 3172480. https://doi.org/10.1155/2018/3172480
- 32. Heeschen, C., et al. (2001) Nicotine Stimulates Angiogenesis and Promotes Tumor Growth and Atherosclerosis. Nature Medicine, 7, 833-839. https://doi.org/10.1038/89961
- 33. Heeschen, C., Weis, M., Aicher, A., Dimmeler, S. and Cooke, J.P. (2002) A Novel Angiogenic Pathway Mediated by Non-Neuronal Nicotinic Acetylcholine Receptors. Journal of Clinical Investigation, 110, 527-536. https://doi.org/10.1172/JCI0214676
- 34. Suleyman, H., et al. (2002) Beneficial Effects of Hippophae rhamnoides L. on Nicotine Induced Oxidative Stress in Rat Blood Compared with Vitamin E. Biological and Pharmaceutical Bulletin, 25, 1133-1136. https://doi.org/10.1248/bpb.25.1133
- 35. Flora, J.W., et al. (2016) Characterization of Potential Impurities and Degradation Products in Electronic Cigarette Formulations and Aerosols. Regulatory Toxicology and Pharmacology, 74, 1-11. https://doi.org/10.1016/j.yrtph.2015.11.009
- 36. Xue, Y., et al. (2010) Venous Plasma Nicotine Correlates of Hormonal Effects of Tobacco Smoking. Pharmacology Biochemistry and Behavior, 95, 209-215. https://doi.org/10.1016/j.pbb.2010.01.007
- 37. Gao, T., et al. (2016) In Vitro Effects of Nicotine on the Non-Small-Cell Lung Cancer Line A549. Journal of Pakistan Medical Association, 66, 368-372.
- 38. Zhang, S., et al. (2017) Evaluation of Whole Cigarette Smoke Induced Oxidative Stress in A549 and BEAS-2B Cells. Environmental Toxicology and Pharmacology, 54, 40-47. https://doi.org/10.1016/j.etap.2017.06.023
- 39. Erat, M., Ciftci, M., Gumustekin, K. and Gul, M. (2007) Effects of Nicotine and Vitamin E on Glutathione Reductase Activity in Some Rat Tissues in Vivo and in Vitro. European Journal of Pharmacology, 554, 92-97. https://doi.org/10.1016/j.ejphar.2006.10.008
- 40. Taylor, M., et al. (2016) E-Cigarette Aerosols Induce Lower Oxidative Stress in Vitro When Compared to Tobacco Smoke. Toxicology Mechanisms and Methods, 26, 465-476. https://doi.org/10.1080/15376516.2016.1222473