Open Journal of Synthesis Theory and Applications
Vol.03 No.02(2014), Article ID:44941,5 pages
10.4236/ojsta.2014.32003
Aromatic Bromination in Concentrated Nitric Acid
Alexander M. Andrievsky1, Vera I. Lomzakova1, Mikhail K. Grachev2, Mikhail V. Gorelik1*
1Research Institute of Organic Intermediates and Dyes (NIOPIK), Moscow, Russia
2Departament of Chemistry, Moscow State Pedagogical University, Moscow, Russia
Email: *gorelik@niopik.ru
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 4 December 2013; revised 3 February 2014; accepted 27 February 2014
ABSTRACT
Action of bromine in concentrated nitric acid allows carrying out mono- and polybromination of moderately deactivated aromatic compounds. 4-Chloronitrobenzene and isophthalic acid turnes into 3-bromo-4-chloronitrobenzene and 5-bromoisophthalic acid at reaction with bromine in concentrated nitric acid at 20˚C whereas in absence of bromine in the same conditions 4-chloro-1, 3-dinitrobenzene and 5-nitroisophthalic acid are formed accordingly. Presence of bromine in concentrated nitric acid changes nitrating capacity to brominating one. Terephthalic acid and phthalic anhydride at heating with bromine in concentrated nitric acid can be transformed to appropriating tetrabromo substituted compounds.
Keywords:
Bromination, Nitration, Aromatic Compounds, 3-Bromo-4-chloronitrobenzene, 5‑Bromoisophthalic Acid, 2,3,5,6-Tetrabromoterephthalic Acid, 3,4,5,6‑Tetrabromophthalic Anhydride

1. Introduction
Aromatic compounds with bromine atom in the ring are of great value in organic chemistry. Bromine is introduced into the aromatic ring as to give desired properties to the final product as, especially, to attain the required reactivity for further syntheses connected with exchange of bromine for various groups. The principal method of bromine introduction into aromatic molecules is electrophilic bromination. It easily proceeds when activating electron-donating substituents are present in the ring but when deactivating electron-withdrawing substituents are present special methods are required [1] .
Earlier we have described bromination of strongly deactivated aromatic compounds by action of bromine in concentrated sulfuric acid in the presence of nitric acid [2] and have discussed competition of bromination and nitration in this media [3] . As participation of nitric acid is of critical importance we have carried out interaction of deactivated aromatics with bromine in pure nitric acid without sulfuric acid. It has appeared that moderately deactivated aromatic compounds which are easily nitrated with concentrated nitric acid are only brominated if concentrated nitric acid contains bromine. Possibility to use treatment with bromine in concentrated nitric acid for synthesis mono- and polybromo substituted aromatics is considered in the present work.
2. Experimental Section
2.1. Reagents and Analytics
Bromine, 99% nitric acid and aromatic substrates were obtained from commercial suppliers and were used without further purification. 1Н and 13C NMR spectra were determined in DMSO-d6 on a Jeol ECX-400 NMR spectrometer, the chemical shifts were reported in δ ppm relative to TMS as internal standard. Correctness of signals reference in 1Н and 13C NMR spectra in addition proved to be true the analysis of two-dimentional spectroscopy HETCOR {1H-13C}. Element analyses are made on the FlashEA 1112HT. Mass spectra were registered on MX-1320 mass spectrometer. IR spectra were recorded on a Perkin-Elmer-598 spectrometer in KBr pellets. Reactions and purity of the formed products were monitored by TLC on Silufol UV-254 plates. The melting points were measured using Boetius apparatus and were uncorrected.
2.2. Syntheses
3-Bromo-4-chloro-1-nitrobenzene (2). 1.58 g (0.01 mol) of 4-chloronitrobenzene 1 was added to mixture of 2.08 g (0.013 mol)) of bromine and 20 ml nitric acid and stirred 4 h at 20˚C until the initial compound 1 was absent. The solution was poured in ice water (100 g), precipitate was filtered off, washed with water and dried. It was obtained 3-bromo-4-chloro-1-nitrobenzene 2, containing small impurity of 3,6-dibromo derivative. Yield 2.22 g (94%), mp 58˚C (EtOH). Found, %: С 30.55, H 1.30, Br 33.51, N 5.85. M+ 236.5. C6H3BrClNO2. Calculated, %: С 30.48, H 1.28, Br 33.79, N 5.92. M+ 236.45.
In the same conditions but without bromine 4‑chloro-1,3-dinitrobenzene 3 was a single product in yield 98%, identical to standard sample.
5-Bromoisophthalic acid (6) and 4,5-dibromoisophthalic acid (7). Isophthalic acid 5 (1.66 g, 0.01 mol) was treated similar to compound 1 for 1 h. It was isolated 1.35 g (55%) of 5‑bromoisophthalic acid 6, mp 280˚C (dec.) (lit. mp 275˚C - 277˚C [4] ). 1H NMR, δ, J, Hz: 8.21 d (2Н, BrСCH, 4J 1.2), 8.37 d (1Н, CCHC, 4J 1.2). NMR 13С, d: 122.5 (СBr), 129.3 (CCHC), 134.0 [CC(O)], 136.2 (BrCCH),165.8 [C(O)]. Found, %: С 39.25, H 2.10, Br 32.50. M+ 245. C8H5BrO4. Calculated, %: С 39.22, H 2.06, Br 32.61. M245.03.
In similar experiment but at duration 22 hours 1.38 g (65%) of 4,5-dibromosophthalic acid 7 was obtained, mp > 230˚C (dec.). 1H NMR, δ, J, Hz: 8.02 d (1Н, BrСCH, 4J 1.8), 8.21 d (1Н, CCHC, 4J 1.2). 13С NMR, d.: 127.1, 127.2 (СBr), 129.2 (BrCCH), 132.1 [BrCCC(O)], 135.7 (CCHC),138.3 [HCCC(O)], 165.4, 167.2 [C(O)]. Found, %: С 29.55, H 1.10, Br 49.15. M+ 324. C8H4Br2O4. Calculated, %: С 29.66, H 1.24, Br 49.33. M+ 323.93.
5-Nitroisophthalic acid (8). Solution of 1.58 g (0.01 mol) of isophthalic acid 5 in 20 ml of nitric acid was stirred 24 h at 20oC and poured into ice water. Precipitate of 5-nitroisophthalic acid 8 (1.50 g, 90%) was filtered off, washed with water and dried. Mp 260˚C (dec.) (lit. mp 255˚C - 258˚C [5] ). 1H NMR, δ, J, Hz: 8.70 dd (1Н, CCHC, 4J 1.4), 8.73 d (2Н, NO2СCH, 4J 1.2). NMR 13С, d: 13С, d, м.д.: 127.8 (CCNO2), 133.7 [CC(O)], 135.5 [(O)CCCHCC(O)] 148.8 (СNO2), 165.4 [C(O)]. Found, %: С 45.45, H 2.48, N 6.53. M+ 211. C8H5NO6. Calculated, %: С 45.51, H 2.39, N 6.63. M+ 211.13.
2,3,5,6-Tetrabromoterephthalic acid (10). 1.66 g, (0.01mol) of terephthalic acid 9 was added in portions to mixture of 6.40 g (0.04 mol) of bromine in 50 ml of nitric acid. The solution is heated 12 h at 50˚C - 55˚C, cooled to 20˚C and after 20 h poured into ice water. Precipitate of tetrabromoterephthalic aid 9 was filtered off, washed and dried, yield 3.20 g (65%), mp >280˚C (lit. mp 300˚C [6] ). NMR 13С, d: 121.9 (СBr), 142.3 [CC(O)], 166.3 [C(O)]. Found, %: С 20.05, Br 66.24. C8H2Br4O4. Calculated, %: С 19.95, Br 66.35.
3,4,5,6-Tetrabromophthalic anhydride (12). Phthalic anhydride 11 (14.8 g, 0.1 mol) was stirred in solution of 32 g (0.2 mol) of bromine in 50 ml of nitric acid 6 h at 50˚C, cooled, 4 g of bromine and 5 ml of nitric acid was added and mixture was heated 8 h at 60˚C, after cooling more 6.2 g of bromine and 5 ml of nitric acid was added and suspension was heated 8 h at 65˚C. Remained bromine was deleted in vacuum. White crystals of 12 (24 g) were separated on the glass filter, washed with 5 ml nitric acid and water. Addition product was collected after dilution of filtrate with water. Total yield of tetrabromophthalic anhydride 12 was 36.6 g (79%); mp 279˚C - 280˚C (toluene) (lit. mp 279.5˚C - 280.5˚C [7] ). NMR 13С, d: 122.3 [CBrCBrCC(O)], 131.2 [CBrCC(O)], 136.6 [CC(O)], 166.4 [C(O)]. Found, %: С 20.86; Br 69.04. M+ 464. C8Br4O3. Calculated, %: С 20.69; Br 68.96. M 463.70.
3-Bromopyromellitic acid (14). 0.64 g (0.0025 mol) of pyromellitic acid 13 and 1.60 g (0.01 mol) of bromine was heated in mixture of 12 ml of concentrated sulfuric acid and 5 ml of nitric acid at 75˚C for 15 h. After dilution with water 3-bromopyromellitic acid 14 was obtained in yield 0.75 g (90%), mp > 270˚C (dec.)). 1H NMR, δ, J, Hz: 8.39 с (1Н, ССНС). NMR 13С, d: 117.0 (CBr), 129.7 [BrCCC(O)], 131.1 (CCHC), 142.7 [HCCC(O)], 165.0, 167.6 [C(O)]. Found, %: С 36.26, H 1.48, Br 24.05. M+ 333. C10H5BrO8. Calculated, %: С 36.06, H 1.51, Br 23.99. M+ 333.05.
3. Results and Discussion
4-Chloronitobezene 1 was chosen as a substrate to characterize new brominating system Br2/HNO3 and to compare with system Br2/H2SO4-HNO3. Treatment of 4‑chloronitobezene 1 with bromine in concentrated nitric acid at room temperature during 4 h leads to 3-bromo-4-chloro-1-nitrobenzene 2 in yield 94%. In the same conditions but in absence of bromine 4-chloro-1,3-dinitrobenzene 3 is formed from 4‑chloronitobezene 1 almost quantitatively (Scheme 1). Bromination does not take place in 60% nitric acid.
In concentrated H2SO4 in the presence HNO3 4-chloronitobezene 1 gives directly 5‑bromo-4-chloro-1,3-dini- trobenzene 4 by nitration at first and following bromination of 4‑chloro-1,3-dinitrobenzene [2] . Dinitrobenzene 3 and other dinitrobenzenes are not brominated with system Br2/HNO3. The fact that compound 1 is brominated with system Br2/HNO3 and is nitrated with system Br2/H2SO4-HNO3 is caused evidently by less nitrating capacity of the first system. Nitric acid exists in sulfuric acid with concenration of 90% and more completely as nitronium cation. Content of nitronium cation in pure nitric acid observed by Raman and IR spectra is about several percents (~3% molar) owing to self-dehydration: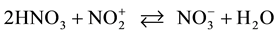 , and it strongly decreases with the addition of 5% water [8] . Nitronium cation is the attacking electrophilic species at aromatic nitration of 4‑chloronitobezene both in concentrated sulfuric acid and in 85% - 95% nitric acid also [9] .
, and it strongly decreases with the addition of 5% water [8] . Nitronium cation is the attacking electrophilic species at aromatic nitration of 4‑chloronitobezene both in concentrated sulfuric acid and in 85% - 95% nitric acid also [9] .
Since bromination in absence of nitric acid does not take place apparently that interaction of bromine with nitronium cation is a key stage in generation of brominating agent. Electron transfer from bromine to nitronium-
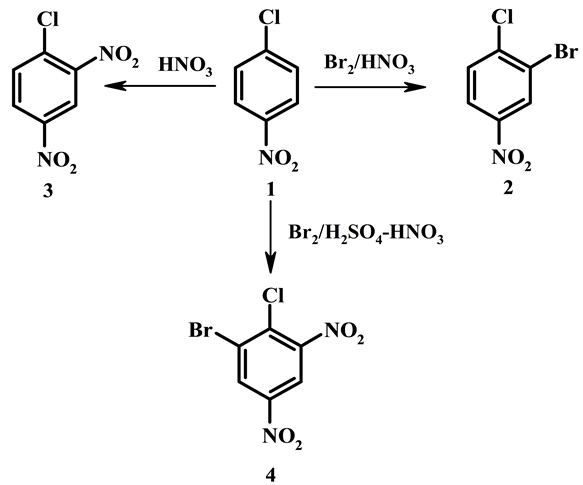
Scheme 1. Reactions of 4-chloronitobezene with bromine in concentrated nitric acid and in concentrated sulfuric acid in the presence of nitric acid.
cation gives brominating species which exceeds an initial nitronium cation on electrophility, and, possible, closed by structure to bromonium cation. Generated brominating agent is so more active than nitrating one that bromination proceed with much greater rate than nitration. Therefore bromination takes place instead of nitration when bromine is present in concentrated nitric acid.
Preparative possibilities of bromination in concentrated nitric acid are demonstrated on an example of benzene dicarboxylic acids. Isophtalic acid 5 turns to 5-bromoisophthalic acid 6 in mild conditions. Compound 6 was isolated after stirring 5 in concentrated nitric acid containing excess of bromine at 20˚C for 1 hours in yield 55%. After 22 hours 4,5-dibromoisophthalic acid 7 was obtained in yield 65%. Treatment of isophthalic acid 5 with concentrated nitric acid at 20˚C in absence of bromine gives 5-nitroisophthalic acid 8 in high yield (Scheme 2). Isophtalic acid 5 is comparable with 4‑chloronitobezene 1 on easiness of bromination and suppression of nitration.
5-Bromoisophthalic acid 6 is of commercial significance as intermediate. The main method for its preparation is bromination of isophthalic acid 5 in oleum at a heat. For example acid 5 is heated with bromine in 10% oleum in sealed tube at 130˚C for 22 hours [10] or in 20% oleum at 102˚C - 107˚C for 6 h in the presence of 2% - 3% of iodine as catalyst [11] . That strongly contrasts with described above process in concentrated nitric acid at room temperature.
Long heating of isophthalic acid 5 with bromine in concentrated HNO3 at 55˚C - 60˚C does not lead to introduction into the ring more than two bromine atoms. Terephthalic acid 9 unlike acid 5 is completely brominated in concentrated HNO3. Heating of terephthalic acid 9 with bromine in concentrated HNO3 at 50˚C - 55˚C for 12 hours gives tetrabromoterephthalic acid 10 in yield 65% (Scheme 3). Earlier this compound was synthesized by bromination of terephthalic acid in 20 % oleum at presence of iodine at 125˚C during 76 hours [12] .
By means of the given method it is possible to introduce four atoms of bromine into molecule of phthalic anhydride. Interaction of phthalic anhydride 11 with bromine in concentrated nitric acid at 50˚C - 65˚C for 22 hours yields 79% of 3,4,5,6-tetrabromophthalic anhydride 12 (Scheme 4). In absence of bromine phthalic anhydride is nitrated to mixture of 3‑nitro and 4-nitro derivatives [13] . Proposed in literature methods for manufacture of tetrabromophthalic anhydride 12 used as flame retarder [14] consists in bromination of phthalic anhydride in strong oleum combined with sulfur dioxide oxidation process [15] [16] .
Accumulation of four carboxylic groups so deactivates benzene ring that prevents bromination in nitric acid. Pyromellitic acid 13 does not changed at interaction with bromine in concentrated HNO3. Bromination is possible to carry out with system Br2/H2SO4-HNO3. Pyromellitic acid 13 turns into 3-bromopyromellitic 14 under stirring in concentrated H2SO4 containing bromine and HNO3 at 75˚C for 15 hours in yield 90% (Scheme 5). Acid 14 was synthesized

Scheme 2.Reactions of isophthalic acid in concentrated nitric acid in the presence and in the absence of bromine.
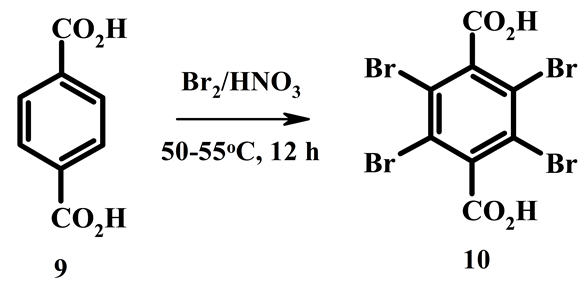
Scheme 3.Reaction of terephthalic acid with bromine in concentrated nitric acid.
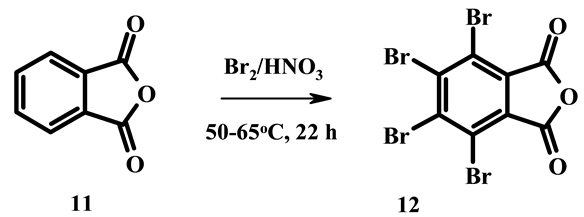
Scheme 4.Reactions of phthalic anhydride with bromine in concentrated nitric acid.
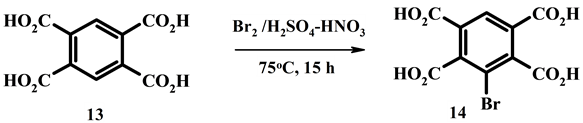
Scheme 5.Reaction of pyromellitic acid in concentrated sulfuric acid in the presence of nitric acid.
earlier by bromination of 1,2,4,5-tetramethylbenzene and following oxidation of methyl groups with potassium permanganate [17] .
4. Conclusion
Thus action of bromine in concentrated nitric acid can be a method for introduction bromine atoms in the ring of moderately deactivated aromatic compounds at rather mild conditions. Brominating agent generated in concentrated nitric acid significantly exceeds on electrophility nitrating agent because of bromination proceeds much more quickly. Presence of bromine in concentrated nitric acid completely suppresses nitrating capacity of the media in relation to moderately deactivated compounds. Strongly deactivated compounds do not react in the given media. Action of bromine in concentrated sulfuric acid in presence of nitric acid is necessary at this case [3] .
References
- Andrievsky, A.M. and Gorelik, M.V. (2011) Bromination of Deactivated Aromatic Compounds. Russian Chemical Reviews, 80, 421-428. http://dx.doi.org/10.1070/RC2011v080n05ABEH004178
- Andrievskii, A.M., Gorelik, M.V., Avidon, S.V. and Al’tman, E.Sh. (1993) Bromination of Deactivated Aromatic Compounds. Russian Journal of Organic Chemistry, 29, 1520-1525.
- Andrievsky, A.M. and Gorelik, M.V. (2013) Competition of Aromatic Bromination and Nitration in Concentrated Sulfuric Acid. Open Journal of Synthesis Theory and Applications, 2, 46-50. http://dx.doi.org/10.4236/ojsta.2013.21005
- Rajesh, K., Somasundaram, M., Saiganesh, R. and Balasubramanian, K.K. (2007) Bromination of Deactivated Aromatics: A Simple and Efficient Method. The Journal of Organic Chemistry, 72, 275-277. http://dx.doi.org/10.1021/jo070477u
- Thiel, W. and Mayer, R. (1986) Synthese und Spektrale Charakterisierung von Blauen Benzenazofarb-stoffen. Journal für Praktische Chemie, 328, S497-S514. http://dx.doi.org/10.1002/prac.19863280406
- Marckwald, W. (1896) Uber die Perhalogenirten Phtalsäuren und des Hexaiodbenzol. Berichte der Deutschen Chemischen Gesellschaft, 29, S1625-S1633. http://dx.doi.org/10.1002/cber.18960290293
- Pratt, D.S. and Young, C.H. (1918) Phthalic Acid Derivatives: Constitution and Colour. XIV. Some Derivatives of Tetrabromophthalimide. Journal of the American Chemical Society, 40, 1415-1425. http://dx.doi.org/10.1021/ja02242a008
- Schofield, K. (1980) Aromatic Nitration. Cambridge University Press, Cambridge, 23-24.
- Veretennikov, E.A., Lebedev, B.A. and Tselinskii, I.V. (2001) Kinetics and Mechanism of p‑Nitrochlorobenzene Nitration with Nitric Acid. Russian Journal of Organic Chemistry, 37, 1451-1454. http://dx.doi.org/10.1023/A:1013408229563
- Nagai, M., Suzuki, H. and Hashiba, I. (Nissan Chem. Ind. Ltd.) (2005) Process for the Preparation of Bromoisophthalic Acid or Derivatives Thereof. US Patent 6855845, Chemical Abstracts, 136, 2002, Article ID: 37401.
- Gelmont, M. and Oren, J. (2002) A New Route for the Preparation of 5-Hydroxyisophthalic Acid. Organic Process Research & Development, 6, 591-596. http://dx.doi.org/10.1021/op0100302
- Knobloch, J.O. and Ramirez, F. (1975) Diacylium Cations from Tetrahaloterephthalic Acids and their Electrophilic Reactivity. The Journal of Organic Chemistry, 40, 1101-1106. http://dx.doi.org/10.1021/jo00896a024
- Odle, R.R. (General Electric Co.) (1986) Nitration of Phthalic Acid or Anhydride. WO 8502615, 1985. Chemical Abstracts, 104, Article ID: 51230.
- Ioffe, D. and Kampf, A. (2007) Bromine, Organic Compounds. In: Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley and Sons, Inc., New York, 340-364.
- Bottelberghe, S.A., McKinnie, B.G., Miller, S.G. and Raposa, D.E. (Ethyl Corporation) (1994) Tetrahalophthalic Anhydride Process. US Patent 5288879, 1994. Chemical Abstracts, 121, Article ID: 10193.
- Harrod, W.B., Hall, T.J., Knight, C.S., Prindle, J.C. and Armstrong, D.M. (2005) Concurrent Sulfur Dioxide Oxidation Process and Its Use in Manufacture of Tetrabromophthalic Anhydride. WO 2005 113430. Chemical Abstracts, 143, Article ID: 479990.
- Holy, P., Sehnal, P., Tichy, M., Zavada, J. and Cısarova, I. (2004) Crystal Structure of 1,10-biphenyl-2,20,3,30-te-tra- carboxylic and 1,10-biphenyl-2,20,3,30,5,50,6,60-octacarboxylic Acids: Solid-State Chiralization and Dissociation. Tetrahedron: Asymmetry, 15, 3805-3810. http://dx.doi.org/10.1016/j.tetasy.2004.10.016
NOTES

*Corresponding author.