Journal of Crystallization Process and Technology
Vol.4 No.1(2014), Article ID:41600,13 pages DOI:10.4236/jcpt.2014.41001
Crystallization of Cellobiohydrolase in the Presence of Cellulose-Degraded Cellobiose: Analysis of Intermolecular Interactions and Association Dynamics
1National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Ibaraki, Japan; 2Maruwa Foods and Biosciences Inc., Tsutsui-cho, Yamatokoriyama, Nara, Japan.
Email: k.onuma@aist.go.jp
Copyright © 2014 Kazuo Onuma et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Kazuo Onuma et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received October 15th, 2013; revised November 15th, 2013; accepted November 22nd, 2013
Keywords: Enzyme Crystallization; Inhibitor; Hamaker Constant; Light Scattering
ABSTRACT
Crystallization of enzymes in presence of impurities is important for clarifying the role of enzymes in natural world. Although it is proposed that impurities inhibit nucleation of enzyme crystallization, details are unclear. In this study, crystallization of cellobiohydrolase from Aspergillus niger was investigated by dynamic and time-resolved static light scattering using cellobiose as an impurity. We aimed to clarify how cellobiose inhibits cellobiohydrolase crystallization and to crystallize cellobiohydrolase in concentrated cellobiose without using seeds. The contribution of attractive forces to total intermolecular interactions of cellobiohydrolase monomers increased with the molar ratio of cellobiose/cellobiohydrolase (R(cb/ce)). Association dynamics of cellobiohydrolase using lithium sulfate, however, showed that the initial aggregation rate decreased with an increase in R(cb/ce). Because binding sites of cellobioses to cellobiohydrolase molecules differed from those for the growth of protein crystals, the binding of cellobioses would increase the chemical potential of the cellobiohydrolase monomers, which gradually reduced supersaturation for growth as the aggregate size increased. This result was in contrast with the conventional idea that cellobiose inhibits the nucleation of cellobiohydrolase crystals. Gentle agitation of cellobiose-containing cellobiohydrolase solutions during sitting-drop vapor-diffusion growth resulted in the growth of cellobiohydrolase single crystals for all R(cb/ce) conditions without using seeds.
1. Introduction
Molecular structure analysis of catalytic enzymes is the key to elucidating roles of proteins and polysaccharides in living organisms. Comprehending the catalytic mechanism and designing artificial catalytic enzymes that act more quickly and efficiently are important issues in pharmaceutical and industrial fields. For example, developing drugs that quickly degrade proteins related to fatal diseases would greatly contribute to healthcare, and reducing the costs of catalysis for biomass production would contribute to the solution of energy problems.
The most powerful and reliable method to analyze the molecular structure of catalytic enzymes is to apply Xray diffraction to high-quality protein single crystals. Although protein crystallization is still problematic, advances in crystallization techniques based on many trials and errors have increased the chances of successful crystallization in pure solutions.
The presence of foreign materials such as impurities and protein-degraded products in the solution used for crystal growth, a common situation, makes crystallization more difficult and sometimes even impossible unless seeds are soaked as the source of secondary nuclei [1,2]. Soaking pure crystal seeds in a solution and using them to grow foreign-material-bound protein crystals is a useful technique for protein crystal growth; however, it is useless if crystallization of pure protein itself is difficult. Therefore, it is important to clarify how these materials inhibit protein crystallization and to find a method to crystallize foreign-material-bound protein crystals without using seeds.
In this study, we focused on the crystallization of cellulase in the presence of cellobiose as an impurity. Cellobiose is a disaccharide that is cleaved by cellobiohydrolase during cellulose hydrolysis. Cellulose is the main component of plant cell walls and the most abundant polysaccharide. Because cellulose can be used to produce bioethanol [3], the enzymatic catalysis of cellulose has been of great interest in the industrial field [4-6]. Cellulases are enzymes that have many structure-related families and that hydrolyze 1,4-b-D-glucosidases such as cellulose. They are usually purified from fungi and bacteria and are characterized as two types, endoglucanases and cellobiohydrolases, depending on the degradation process of cellulose. Cellobiohydrolases processively act from the reducing to non-reducing end of the cellulose chain, producing cellobiose molecules. Most cellulases have two domains: a large catalytic core domain and a small cellulose-binding module domain connected by a flexible linker.
There have been many crystallization and structure analysis studies both for endoglucanases [7-11] and cellobiohydrolases [12-17] purified by several sources. These studies mainly focus on the structure of the catalytic core domain. Crystallization in the presence of foreign materials has also been studied using the seeding technique to obtain crystals [18-20]. Representative structural analysis for binding between endoglucanase (AaCel9A) [19] and cellobiose or cellobiohydrolase (Pc_Cel7D) [20] and cellobiose showed that for Aa-Cel9A and Pc_Cel7D, cellobiose binds in three glucosyl-binding subsites (−1, −2, and +1) and two glucosyl-binding subsites (+1 and +2), respectively. Binding sites in Pc_Cel7D are close to the exit of the cellulose-binding cleft. Inclusive of these reports, no investigation has been performed on crystallization dynamics in the presence of cellobiose.
There has been a common assumption among researchers that cellobiose inhibits the association of protein molecules by increasing the energy barrier of nucleation in a similar manner to that observed for protein oligomers [21]. Seed soaking produces secondary nuclei that remove the nucleation barrier, thereby enabling crystallization.
One aim of this study was to investigate the role of cellobiose in cellobiohydrolase crystallization and determine whether it actually inhibits the nucleation of protein crystallization. Using custom-made light scattering equipment, we compared the intermolecular interaction of cellobiose-bound cellobiohydrolases with that of pure cellobiohydrolase monomers and clarified the association process of proteins under various cellobiose concentration conditions. Then, on the basis of the analysis of the light scattering data, we developed a simple but effective technique for quickly growing cellobiohydrolase crystals in cellobiose-containing solutions without using seeds, for which there are no reports of successful studies to our knowledge.
2. Experimental Section
2.1. Purification of Cellobiohydrolase and Solution Conditions
Purification of full-length cellobiohydrolase (possessing two domains and sugar fragments bound to a protein molecule) by chromatography was carried out at 4˚C. Cellobiohydrolase powder prepared from Aspergillus niger was purchased from Wako Pure Chemical Industries, Ltd. One gram of the powder was dissolved in a 30 mL solution containing 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)–NaOH buffer at a pH of 7.0. The protein solution was purified in consecutive steps by anion exchange chromatography (Q-Sepharose FF column with two buffers (20 mM HEPS-NaOH + 2 mM CaCl2 and 20 mM HEPES-NaOH + 2 mM CaCl2 + 1 M NaCl at pH of 7.0); GE Healthcare), hydrophobic interaction chromatography (Phenyl TOYOPEARL 650 s column with two buffers (20 mM succinic acid + 2 M ammonium sulfate and 20 mM succinic acid at pH of 6.0); TOSO), and anion exchange chromatography (QSepharose HP column with two buffers (20 mM HEPESNaOH + 2 mM CaCl2 + 0.0008% NaN3 and 20 mM HEPES-NaOH + 2 mM CaCl2 + 0.0008% NaN3 + 1 M NaCl at pH of 7.0; GE Healthcare). The fractions corresponding to cellobiohydrolase were collected, and their purity was analyzed using SDS-PAGE, native-PAGE, and high-performance liquid chromatography with a SuperQ- 5PW column. After chromatography, the purified protein solution was dialyzed with a buffer containing 20 mM HEPES-NaOH and 2 mM CaCl2 at a pH of 7.0 at 20˚C. Hereafter, we called this buffer the standard buffer. The protein solution was concentrated using an AMICON ultrafiltration device with a YM-10 membrane.
A cellobiose solution was prepared by dissolving cellobiose powder (Sigma Aldrich Inc., molecular weight 342.3) in the standard buffer at a concentration of 10 mg/mL. When the cellobiohydrolase solution containing cellobiose was prepared, the cellobiose solution was added so that the molar ratio of cellobiose/cellobiohydrolase, R(cb/ce), in the mixed solution ranged from 0 to 10. We prepared solutions with R(cb/ce) = 0 (pure cellobiohydrolase), 1, 2, 3, 5, 7, and 10.
Light scattering and crystallization experiments were performed at 20˚C. The crystallization reagent used in time-resolved static light scattering (SLS) measurements and in cellobiohydrolase crystallization by sitting-drop vapor diffusion was 2.5 M lithium sulfate dissolved in 200 mM sodium citrate buffered at a pH of 6.0 using citric acid. We used a pH of 6.0 to stabilize the solution pH and thereby increase the maximum concentration of lithium sulfate. Owing to sodium citrate, a solution pH of 7.0 was unstable; thus, the maximum lithium sulfate concentration in the solution was limited to ~1.8 M. Combining lithium sulfate, sodium citrate, and a low concentration of calcium at around neutral pH has been the only successful approach to grow high-quality cellobiohydrolase single crystals from Aspergillus niger at 20˚C (Inaka et al., unpublished data). To compare the results of this study with those of a previous crystallization study of pure cellobiohydrolase, we used exactly the same chemicals.
2.2. Dynamic and Time-Resolved Static Light Scattering Measurements
Dynamic light scattering (DLS) measurements for the protein solutions [22-26] containing cellobiose at each R(cb/ce) were conducted using a multi-angle DLS system with a 100 mW solid-state laser (wavelength lW = 533 nm) as the light source. After the cellobiose solution was mixed with the protein solution, the sample was placed in an incubator set at 20˚C ± 0.5˚C for ~1 h; it was then filtered using a 0.22 µm pore membrane prior to DLS measurements. It is generally considered that cellobiose stably binds to cellobiohydrolase molecules within a few minutes. The sample solution (~150 mL) was placed in a glass tube with an inner diameter of 5 mm, the type of tube conventionally used for nuclear magnetic resonance measurements. The second autocorrelation functions were simultaneously measured at scattering angle q from 30˚ to 100˚ with a step of 10˚ to determine the angular dependence. The typical elapsed time for each measurement was 120 s. CONTIN fitting [27] was used to analyze the autocorrelation function at each scattering angle to calculate the decay time, t. The relationship between  and diffusion coefficient D was estimated using
and diffusion coefficient D was estimated using
 (1)
(1)
where q is the scattering vector proportional to scattering angle q ( ).
).
Because D measured at a finite protein concentration is an apparent value modified by the intermolecular interactions, the actual translational diffusion coefficient D0 at the limit of the zero protein concentration, where the protein molecules freely diffuse, was estimated by measuring the dependence of D on the concentration. The intermolecular interaction parameter, l, was estimated using
 , (2)
, (2)
where f is the volume fraction expressed using the partial molar volume of the monomers, n, and the protein concentration, . We used 0.73 × 10−3 mL/mg for n, which is the typical value for protein molecules.
. We used 0.73 × 10−3 mL/mg for n, which is the typical value for protein molecules.
The solvent viscosity required to convert the translational diffusion coefficient into the hydrodynamic radius of molecules, rH, was measured using a vibration-type viscometer (SV-10, A & D Co., Ltd.).
The molecular weight of cellobiohydrolase (without cellobiose) in the standard buffer was measured using a fixed-angle SLS detector (Zetasizer Nano, Sysmexs Co., Ltd.) with a 1 mW He-Ne laser (lW = 632.8 nm) as the light source. The protein solutions were filtered before measurements in the same manner as for the DLS measurements. Data were obtained at a scattering angle of 90˚ and the molecular weight was estimated by extrapolating the inverse of the solution scattering intensity to the zero protein concentration using the Debye plot,
 , (3)
, (3)
where DR is the excess scattering intensity of the protein solution to that of the solvent (excess Rayleigh ratio). K is a constant including the refractive index increment of the solution against the protein concentration, dn/dc, Mw is the molecular weight of the measured particles, and A2 is the second virial coefficient of particles. The dn/dc value was measured for calculating the absolute molecular weight using an Abbe-type refractometer (RX-7000a, Atago Co., Ltd.) with an accuracy of 10−4.
All DLS and SLS measurements were repeated 10 times for each condition, and average values with their standard deviations (corresponding to error bars) were calculated.
The association of cellobiohydrolase molecules after the protein solution (containing cellobiose) was mixed with the crystallization buffer at a volume ratio of 1:1 was observed using a multi-angle SLS detector [28-32]. The concentration of cellobiohydrolase in the mixed solution was 7 mg/mL and that of lithium sulfate was 1.25 M. The scattering intensities at q from 20˚ to 150˚ with a step of 1˚ were simultaneously measured over time at intervals of 3 s for each R(cb/ce) condition. The molecular weights and gyration radius of the aggregates were calculated by applying the Zimm-square-root equation to the data:
 , (4)
, (4)
where Rg is the gyration radius of the aggregates.
Since the extrapolation of the plot to the limit of the zero protein concentration was impossible, the calculated molecular weight was the apparent value. The contribution of second term A2c was neglected, as usual in timeresolved SLS measurements, because the contribution of the first term is much higher.
The time required to prepare the solutions before starting the measurements affects the results of time-resolved SLS measurements. Thus, we strictly set the time for mixing the protein solution with the crystallization solution to 10 s under constant mixing speed conditions. This lengthy mixing resulted in a uniform concentration distribution of highly viscous lithium sulfate in the mixed solution. The time required to place the sample in the SLS device after mixing was 15 s. The protein and the crystallization solutions were placed in the incubator, which was set to 20˚C ± 0.5˚C, for ~1 h and were removed just before they were mixed. Both solutions were filtered using a 0.22 µm pore membrane before mixing and were not filtered after mixing.
2.3. Capillary Electrophoresis Measurement
The surface potential y0 of the cellobiohydrolase monomers was estimated using the capillary electrophoresis measuring system CAPI-3300 (Otsuka Electronics Co., Ltd.). The electrophoretic mobility of the protein molecules was measured in the standard buffer for each R(cb/ce) condition. A silica capillary with a total length of 50 cm (38 cm effective length) and an inside diameter of 75 µm was used for the measurements, which were performed with a 20 kV applied voltage. The protein solution was filtered using a 0.22 µm pore membrane before the measurement. The migrating particles were detected using the absorbance at a wavelength of 200 nm.
The relationship of y0 to the electrophoretic mobility of particles m is given by Henry’s equation:
 (5)
(5)
 (6)
(6)
where z is the zeta potential, which was assumed to be equal to y0, h denotes the viscosity of the medium, e is the dielectric constant of the medium, e0 is the permittivity of free space, a is the particle radius, and k is the screening parameter, which is given by

 (7)
(7)
where e is the elemental charge, kB is the Boltzmann constant, T is the absolute temperature, nj is the number density (m−3), and zj is the ion valence of the jth ion.
The surface charge, Z, of cellobiohydrolase monomers in the standard buffer was calculated using
 , (8)
, (8)
where a was assumed to be equal to rH.
All capillary electrophoresis measurements were repeated 10 times, and the average values with their standard deviations were calculated.
2.4. Cellobiohydrolase Crystallization Using Sitting-Drop Vapor Diffusion
Cellobiohydrolase crystallization was performed by sitting-drop vapor diffusion. The solutions used for the time-resolved SLS measurements were collected after the measurements were completed (~3 h after the preparation of the initial protein solution containing the crystallization reagent), and portions of these were used as mother drops in sitting-drop vapor diffusion. The volume of each drop was ~10 mL. The outer solution used for the diffusion was the crystallization solution (2.5 M lithium sulfate and 200 mM sodium citrate). Crystallization behavior was observed using a transmission-type optical microscope (BX-60, Olympus Co., Ltd.).
3. Results and Discussion
3.1. Protein Purification, Hydrodynamic Radius, and Molecular Weight of Cellobiohydrolase in the Absence of Cellobiose
Figure 1(a) shows a typical SDS-PAGE pattern of purified cellobiohydrolase. A single narrow band corresponding to an Mw of ~56 kDa was observed. A corresponding impurity band such as a covalent-bonded dimer was not observed.
Figure 1(b) shows an example of the CONTIN plot of the decay time distribution, proportional to the diffusion coefficient, obtained by analyzing the autocorrelation function data, which were measured for a pure cellobiohydrolase solution using DLS. The protein concentration was 7 mg/mL. The decay times at three representative scattering angles (30˚, 60˚, and 100˚) are shown in the figure. The peaks for all tested protein concentrations had a monomodal distribution independent of the scattering angles.

Figure 1. (a) SDS-PAGE image of purified cellobiohydrolase. (b) Decay time distribution of cellobiohydrolase molecules in standard buffer measured using DLS. Representative results at three scattering angles are shown. (c) Relationship between the diffusion coefficient and protein concentration. The positive slope of the correlation line indicates that total intermolecular interaction between molecules is repulsive. (d) Debye plot of cellobiohydrolase molecules measured in standard buffer using SLS. Mw was ~52 kDa, which corresponds to that of cellobiohydrolase monomers.
The dependence of the diffusion coefficient on the protein concentration calculated using the detected monomodal peaks was linear with a positive slope indicating that the total intermolecular interaction was repulsive (Figure 1(c)). The errors of all data were within 2%. The y-axis intercept of the correlated line showed that the average D0 of the cellobiohydrolase molecules was (69.4 ± 1.4) × 10−12 m2/s, which was converted into the rH, of the molecules (rH = 3.01 ± 0.06 nm) after correcting for the solvent viscosity (1.03 cP).
Kc/DR dependence on the protein concentration was determined by SLS measurement (Figure 1(d)). The errors of data were within 5%. The y-axis intercept of the correlated line showed that the average Mw of cellobiohydrolase in the standard buffer was 52 ± 2 kDa, which corresponds to that of monomeric cellobiohydrolase including sugar chains around protein molecules. Consistent with the DLS result, positive A2 proportional to the inclination of the correlation line indicates that a repulsive force is dominantly operated between cellobiohydrolase monomers.
3.2. Change in the Intermolecular Interaction of Cellobiohydrolase Monomers at Different R(cb/ce)
Figure 2(a) shows the relationship between the D of cellobiohydrolase monomers and the cellobiohydrolase concentration at each R(cb/ce). Five representative conditions, R(cb/ce) = 0, 1, 3, 5, and 10, were plotted for ease of viewing. The relationship was linear for each condition. The y-axis intercepts of correlated lines were deviated within 2% compared with that of a pure cellobiohydrolase solution, indicating that the D0 of monomers is independent of the cellobiose concentration.
Figure 2(b) shows the D/D0 depending on f at R(cb/ce) = 0, 1, 3, 5, and 10. The l value was calculated using the
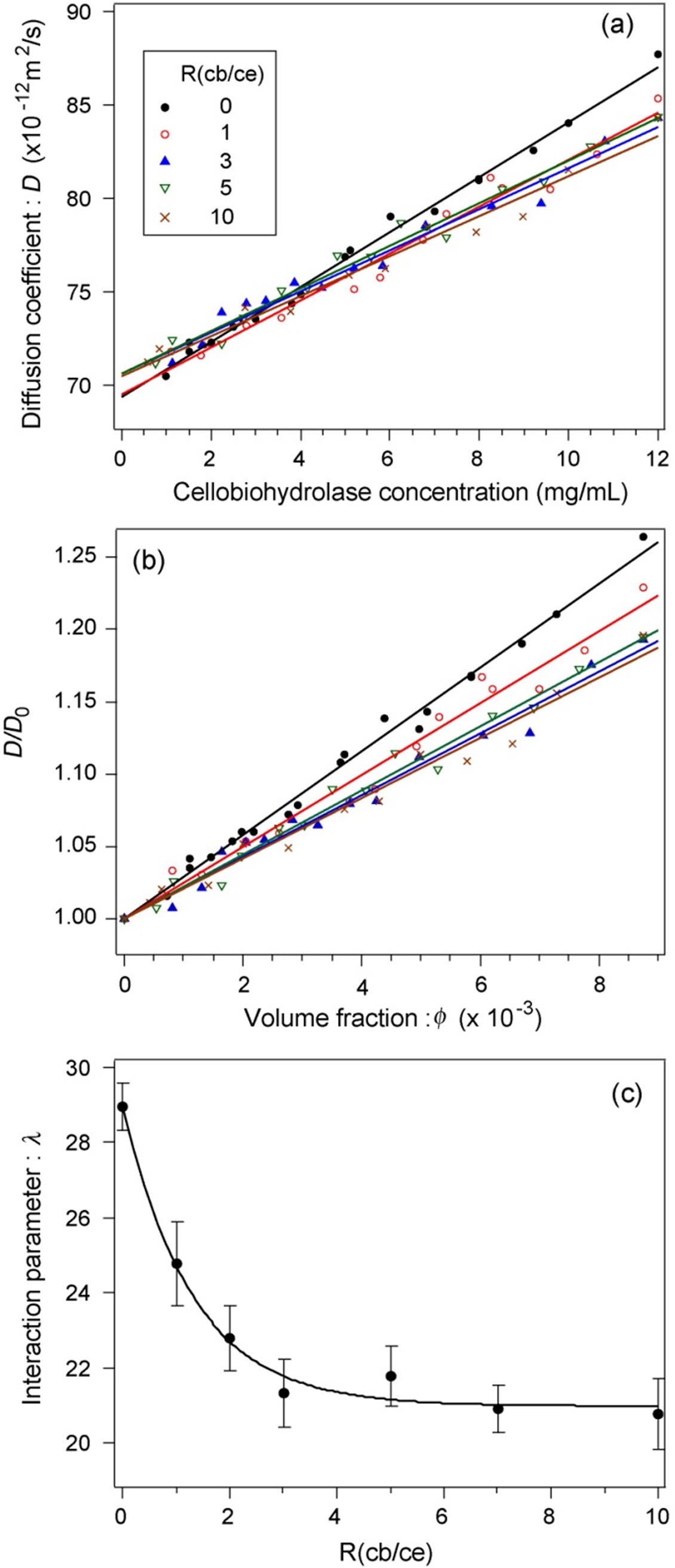
Figure 2. (a) Relationship between the diffusion coefficient of cellobiohydrolase monomers and cellobiohydrolase concentrations at various R(cb/ce). (b) Normalized diffusion coefficients of cellobiohydrolase monomers. The contribution of the attractive forces to the total intermolecular interaction increased with the cellobiose concentration. (c) Dependence of the interaction parameter on R(cb/ce).
slope of each correlated line.
Figure 2(c) shows the relationship between l and R(cb/ce). The l exponentially decreased with an increase in R(cb/ce) from 0 to 3 and was virtually constant from 3 to 10, indicating that the contribution of attractive forces to total intermolecular interactions increased when cellobiose binds to cellobiohydrolase. Both the change in the surface charge and that in the Hamaker constant of cellobiohydrolase monomers could cause this attraction shift. When the binding of cellobiose increases the average surface charge of monomers, the Hamaker constant should increase to shift the molecular interaction to attractive. A change in one or both parameters is of course probable. We thus measured the average surface charge of monomers by capillary electrophoresis.
3.3. Surface Charge of Cellobiohydrolase Monomers
Figure 3(a) shows an example of the migration spectrum (absorbance of migrating particles versus time) for the pure cellobiohydrolase solution at a concentration of 2 mg/mL. The small peak at around t = 4.5 min corresponds to the electro-osmotic flow and was used as a reference peak for calculating the electrophoretic mobility m of cellobiohydrolase monomers. The main peak at around t = 11 min corresponds to the monomers and is smooth without any shoulders. The spectra for all R(cb/ce) conditions were similar. Previous capillary electrophoresis measurements for other protein solutions showed that the m of the monomers depends on the protein concentration, although the reason remains unclear [26,31]. We thus measured the concentration dependence of m for all solutions and estimated the actual m by extrapolation of the correlation line to the zero protein concentration.
Figures 3(b) and (c) show the relationship between m calculated using the migration spectrum and the protein concentration for each R(cb/ce) condition. The errors of all data were within 0.7%. The concentration dependences of m at various R(cb/ce) showed similar trends; they slightly increased with a decrease in the concentration. No particular dependence was observed between the slope of the correlation line and R(cb/ce).
Figure 3(d) shows the relationship between Z and R(cb/ce). The solid line indicates the average Z (−12.5), and the two dashed lines show the upper and lower limits (±1% from the average). The Z value was independent of R(cb/ce), indicating that the binding of the cellobiose molecule(s) to a protein monomer does not change the average surface charge.
The X-ray structure analysis of cellobiohydrolase single crystals has shown that the molecules have an ellipsoidal form and are bent around the center, resulting in a C-shape (Inaka et al., unpublished data). Here the cellobiose molecules were bound to the cleft around the sur-

Figure 3. (a) Electrophoretic migration spectrum for pure cellobiohydrolase in standard buffer. (b) Electrophoretic mobility versus cellobiohydrolase concentration for R(cb/ce) = 0, 1, 2, and 3 measured in standard buffer. (c) Electrophoretic mobility versus cellobiohydrolase concentration for R(cb/ce) = 5, 7, and 10. (d) Surface charge versus R(cb/ce).
face of the central region, indicating that the effect of cellobiose binding on the surface charge of cellobiohydrolase molecules is essentially small. The results plotted in Figure 3(d) are consistent with those of the X-ray structure analysis.
3.4. Hamaker Constant of Cellobiohydrolase Monomers
The Hamaker constant of cellobiohydrolase monomers in the standard buffer at each R(cb/ce) was estimated using the results of the intermolecular interaction parameter and surface charge [22-24,26,31,32]. The Derjaguin-Landau-Verwey-Overbeek model [33] was used to calculate the constant. Since there was no polymer in the solution that would create an attractive force owing to osmotic depletion and no evidence of specific salt bridges between monomers, as shown by X-ray structure analysis, we assumed the simplest model for intermolecular interactions. In this model, there is competition between electrostatic repulsion, Ues, caused by the surface charge of the monomers and van der Waals attraction, Uvdw, of the monomers. The radial distribution function of monomers g(r) expresses the intermolecular interaction potential of mean force U(r) as
 . (9)
. (9)
The relationship between g(r) and interaction parameter l is described elsewhere [26]. The total potential is expressed as , and
, and
 (10)
(10)
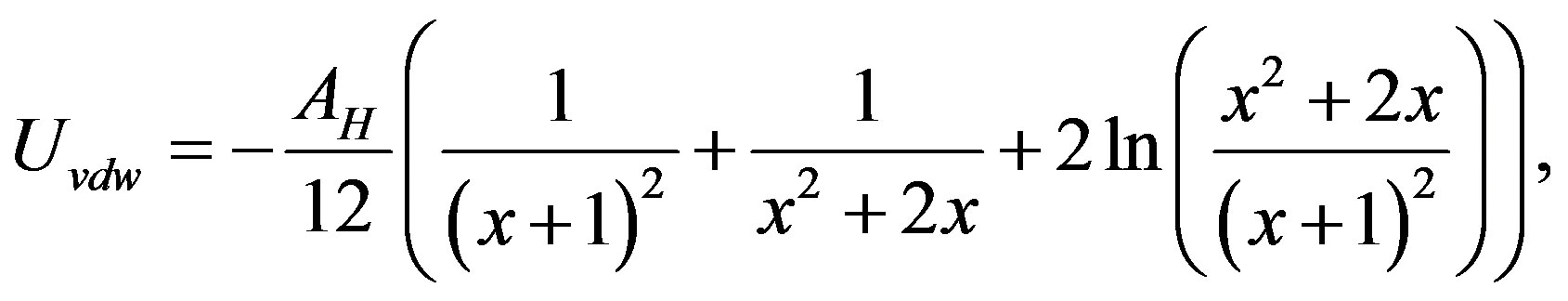 (11)
(11)
where  (a corresponds to rH), and AH is the Hamaker constant of the monomers. The expression of the attractive potential between monomers (Equation
(a corresponds to rH), and AH is the Hamaker constant of the monomers. The expression of the attractive potential between monomers (Equation
(11)) is the most primitive one. Additional attractive forces originating from hydration force [34,35] and hydrophobic molecular interaction [36,37] might also be operating. The effect of these attractive forces is therefore included in calculated AH.
Figure 4(a) shows the (AH, Z) diagram for four representative R(cb/ce) conditions (0, 1, 3, and 10). AH was estimated using the results plotted in Figure 3(d). The relationship between AH and R(cb/ce) is plotted in Figure 4(b): AH quickly increased when R(cb/ce) was increased from 0 to 3 and then was virtually constant when it was increased from 3 to 10. The difference in AH between R(cb/ce) = 0 and 10 was ~8%, which is sufficient to affect the association behavior. The error in each AH was attributed to the lower and upper limits of Z against the average Z value (Figure 3(d)). Figure 4(b) indicates that the binding of cellobiose essentially increases the attractive interaction between monomers; therefore, aggregation should be faster when the cellobiose concentration is higher. However, the actual crystallization behavior did not match the expected behavior. As described in the introduction and discussed later, cellobiohydrolase crystallization was inhibited when cellobiose was added to the solution. We addressed this inconsistency by investigating the association of cellobiohydrolase with and without cellobiose. There have been no quantitative reports on the growth rate of cellobiohydrolase aggregates during the initial stage of crystallization.
3.5. Association Dynamics of Cellobiohydrolase Depending on R(cb/ce)
Time-resolved SLS measurements revealed aggregate formation when the crystallization solution was mixed with the standard buffer without cellobiohydrolase. Because both the crystallization and buffer solutions were stable (no aggregates were observed in either solution), this finding suggests that part of lithium sulfate reacted with HEPES. The scattering intensity of the background solvent is generally assumed to be constant in SLS experiments; however, this assumption could not be applied to the present measurements. Therefore, we monitored aggregation that occurred when the crystallization solution was initially mixed with standard buffer and then directly subtracted the scattering intensity at each scattering angle from that measured for the protein solutions mixed with the crystallization one over time.
Figure 5(a) shows the  versus
versus  plot for the aggregates that appeared in the crystallization solution mixed with the standard buffer at different association periods. The relationship was linear for all
plot for the aggregates that appeared in the crystallization solution mixed with the standard buffer at different association periods. The relationship was linear for all  ranges less than ~2.5 min. The
ranges less than ~2.5 min. The 
value at high scattering angles gradually deviated from linear dependence over time. The data for  < 0.4 could be linearly fitted up to 13 min; however, the scattering angle range quickly narrowed after 13 min. This was due to the development of form anisotropy of the aggregates over time. Fitting of the data assuming an aggregate form such as a simple rod or disk was found to be useless. The intersection of the correlated curve dispersed over time, which inhibited accurate estimations of the change in Mw (inverse of y-axis intercept of the correlation curve) and in Rg (tangent of the correlation curve at q = 0). Thus, we limited the analysis of the time-resolved SLS data to the initial 10 min and for the range
< 0.4 could be linearly fitted up to 13 min; however, the scattering angle range quickly narrowed after 13 min. This was due to the development of form anisotropy of the aggregates over time. Fitting of the data assuming an aggregate form such as a simple rod or disk was found to be useless. The intersection of the correlated curve dispersed over time, which inhibited accurate estimations of the change in Mw (inverse of y-axis intercept of the correlation curve) and in Rg (tangent of the correlation curve at q = 0). Thus, we limited the analysis of the time-resolved SLS data to the initial 10 min and for the range  (corresponding to 60˚), where the data were linearly correlated not only for this solution but also for the protein solutions.
(corresponding to 60˚), where the data were linearly correlated not only for this solution but also for the protein solutions.
Figure 5(b) shows the distribution of decay times of the aggregates measured ~2 h after mixing the crystallization solution with the standard buffer (DLS data). A

Figure 4. (a) Diagram for the Hamaker constant of cellobiohydrolase monomers versus the average surface charge depending on R(cb/ce). Four representative conditions for R(cb/ce) are plotted. (b) Change in the Hamaker constant of monomers with the change in R(cb/ce).

Figure 5. (a) (Kc/ΔR)1/2 versus sin2(q/2) plot depending on the observation time for aggregates observed in the reference solution. Solid lines show the range in which the data are fitted by linear correlation. Dashed lines are extrapolations of solid lines. (b) Decay time distribution of aggregates in the reference solution after 2 h of solution preparation (DLS data).
monomodal peak was observed for each scattering angle. The decay times were converted into an apparent hydrodynamic radius of ~400 mm using Equation (1). Further growth of the aggregates over time was not observed.
Figure 6(a) shows the (Kc/DR)1/2 versus  plot for the aggregates observed in pure cellobiohydrolase solutions mixed with crystallization solutions at a volume ratio of 1:1. All-range linear fitting was already violated at t = 0, and the linear-fitted range quickly narrowed after 1 min. We obtained
plot for the aggregates observed in pure cellobiohydrolase solutions mixed with crystallization solutions at a volume ratio of 1:1. All-range linear fitting was already violated at t = 0, and the linear-fitted range quickly narrowed after 1 min. We obtained  at 10 min, and the change in the plot shape was subsequently gentle. These behaviors were similarly observed for the other protein solutions containing cellobiose (R(cb/ce) = 1 - 10), although the period in which the linear-fitted range was limited to
at 10 min, and the change in the plot shape was subsequently gentle. These behaviors were similarly observed for the other protein solutions containing cellobiose (R(cb/ce) = 1 - 10), although the period in which the linear-fitted range was limited to  varied with the cellobiose concentration.
varied with the cellobiose concentration.
Figures 6(b) and (c) show the change in Mw and Rg, respectively, of the cellobiohydrolase aggregates over time depending on R(cb/ce) in the 0 - 3 range after subtraction of the scattering intensity change for the reference solution. The apparent Mw linearly increased over the initial 10 min. The initial growth rate of the aggregates was calculated using the slope of the fitting line in Figure 6(b). In the solutions with , the calculated Mw was widely dispersed (more than 100%) depending on the measurements even though the conditions were fixed. The initial growth rate of the aggregates was sometimes comparable with that for R(cb/ce) = 3; while at other times, it was higher than that for R(cb/ce) = 1. The reason is unclear. X-ray structure analysis showed multiple cellobiose-binding sites on the monomers, and the maximum number of cellobiose molecules bound to a monomer would be five. The light scattering data suggest that some of the bound cellobiose was unstable. Free cellobiose molecules interact with lithium sulfate that forms aggregates, which heterogeneously assists the aggregation of cellobiohydrolase.
, the calculated Mw was widely dispersed (more than 100%) depending on the measurements even though the conditions were fixed. The initial growth rate of the aggregates was sometimes comparable with that for R(cb/ce) = 3; while at other times, it was higher than that for R(cb/ce) = 1. The reason is unclear. X-ray structure analysis showed multiple cellobiose-binding sites on the monomers, and the maximum number of cellobiose molecules bound to a monomer would be five. The light scattering data suggest that some of the bound cellobiose was unstable. Free cellobiose molecules interact with lithium sulfate that forms aggregates, which heterogeneously assists the aggregation of cellobiohydrolase.
Figures 6(b) and (c) evidently indicate that cellobiohydrolase molecules aggregate even if there is a high concentration of cellobiose in the solution. The previous understanding that cellobiose inhibits the nucleation of cellobiohydrolase crystals is thus incorrect. Submicrometer-sized aggregates are stably formed in the solution.
No crystal or amorphous precipitation was observed in any of the R(cb/ce) solutions during the observation period (~30 days). Cellobiohydrolase crystallization using a batch method with lithium sulfate as a crystallization reagent was impossible even in a pure solution. We tried to increase the concentration of lithium sulfate (up to 2 M after mixing with the protein solution) or cellobiohydrolase (up to 15 mg/mL after mixing with 2.5 M lithium sulfate); however, crystals did not precipitate in the solutions. Hangingor sitting-drop vapor diffusion to increase the lithium sulfate concentration was necessary to grow crystals.
Figure 6(d) shows the initial growth rate of cellobiohydrolase aggregates versus R(cb/ce), as estimated using the data plotted in Figure 6(b) (change in mass weight over time). The growth rate exponentially decreased with an increase in R(cb/ce). This is consistent with the crystal growth behavior observed for the cellobiose-containing solution in which the crystallization of cellobiohydrolase was inhibited strongly.
A question arises about why the growth rate decreased while the attractive intermolecular interaction increased with R(cb/ce) (Figure 4). Because the cellobiose bound to cellobiohydrolase molecules at fixed sites is different from the binding sites between protein molecules during crystallization (Inaka et al., X-ray structure analysis, unpublished data), a reasonable explanation of growth inhibition is that cellobiose binding caused a slight increase

Figure 6. (a) (Kc/ΔR)1/2 versus sin2(q/2) plot depending on the observation time for cellobiohydrolase aggregates in a pure solution. (b) Change in the apparent molecular weight of aggregates over time for solutions with R(cb/ce) = 0 (black), 1 (red), 2 (blue), and 3 (purple). (c) Change in the gyration radius of aggregates over time. Colors are the same as in (b). (d) Initial growth rate of aggregates calculated the data in (a) vs. R(cb/ce). The growth rate quickly decreased with an increase in R(cb/ce).
in the chemical potential of the cellobiohydrolase molecules, which became more effective with the growth of aggregates. This potential increase, of course, affects the chemical potential of the solution phase; however, it could be neglected because the density of protein molecules per unit volume is far smaller in the solution than that in the solid phase (aggregates). The actual driving force for the growth of aggregates, which is the difference of the potential between the liquid phase and that of the solid phase, therefore, decreased with the aggregate size. We suppose that cellobiose binding would induce the faint strain into cellobiohydrolase molecules, which caused the potential increase in molecules. This strain in single molecules would be very weak because X-ray structure analysis did not detect any molecular distortion in the cellobiose-bound cellobiohydrolase compared to pure celobiohydrolase, which is consistent in the case of endoglucanase [19]. The total potential increase in the condensed solid phase became evident with the growth of aggregates, which inhibited the further growth to macroscopic crystals. The change in the morphology of cellobiose-bound cellobiohydrolase crystals (see next section) compared with that of pure cellobiohydrolase crystals suggests that the binding between protein molecules is modulated.
These observations and discussion help clarify the reason for the growth of cellobiohydrolase crystals in high R(cb/ce) solutions. Because aggregates formed in cellobiose-containing solutions and the intermolecular interactions of cellobiose-bound protein molecules were more attractive than those of pure cellobiohydrolase molecules, increasing the sticking (contact) probability between aggregates should result in crystal growth, even though the driving force for growth is weaker in cellobiose-containing solutions.
3.6. Crystallization of Cellobiohydrolase under High R(cb/ce) Conditions
Figure 7(a) shows an optical microscope image of single crystals grown in pure cellobiohydrolase solutions by sitting-drop vapor diffusion. Rugby-ball-shaped plate crystals were observed after 1 day of drop setting. This image was taken after 3 days of setting. Figure 7(b) shows the crystals grown in the R(cb/ce) = 1 solution. Tiny crystals were observed after 3 days of setting. The image was taken 5 days after the first crystal was observed. The morphology of the crystals differed from that of pure crystals. The rounded side faces of the crystals changed to polygonal faces, and the aspect ratio of the crystals decreased. When R(cb/ce) exceeded 2, the crystals were not observed during the observation period (~30 days).
After the concentration of lithium sulfate in the drops had increased sufficiently (~1 day after drop setting), we applied gentle agitation to the drops during crystallization to increase the sticking probability between the aggregates. Sucking and ejecting of the solution (~3 µL) three to five times at a rate of 1 Hz using a micropipette resulted in cellobiohydrolase crystallization for all R(cb/ ce) conditions. Figure 7(c) shows a crystal grown using this agitation method for R(cb/ce) = 3. The crystals were observed within 1 day after applying agitation. This image was taken 3 days after the first crystal was observed. Note that the crystal morphology is similar to that of R(cb/ce) = 1. The time necessary to find tiny crystals was similar even for R(cb/ce) = 10, and the morphology of the crystals showed no evident change in this solution, as shown in Figure 7(d). More vigorous agitation (sucking and ejecting ten times) resulted in the precipitation of many microcrystals, as shown in Figure 7(e). This image was taken after 1 day of agitation for R(cb/ce) = 2.
4. Conclusion
Cellobiohydrolase crystallization in the presence of the cellulose-degraded product, cellobiose, was investigated using light scattering on the basis of the analysis of intermolecular interactions and association dynamics. The

Figure 7. (a) Cellobiohydrolase crystals precipitated in a pure solution using sitting-drop vapor diffusion. Tiny crystals were observed after setting for 1 day. The image was taken after setting for 3 days. (b) Cellobiohydrolase crystals naturally precipitated in R(cb/ce) = 1 solutions. The image was taken after 5 days when the precipitation of crystals was observed. (c) Cellobiohydrolase crystal precipitated in R(cb/ce) = 3 solutions. Gentle agitation was applied to the drop after setting for 1 day resulting in crystallization just after 1 day. The image was taken after 3 days when the precipitation of crystals was observed. (d) Cellobiohydrolase crystals precipitated in R(cb/ce) = 10 solutions. Gentle agitation was applied to the drop. The image was taken after 1 day of agitation application. (e) Precipitation of many small cellobiohydrolase crystals in R(cb/ce) = 2 solutions following the application of hard agitation. The image was taken after 1 day of agitation application.
interaction of cellobiohydrolase monomers shifted to be attractive when cellobiose molecules were bound to them owing to an increase in the Hamaker constant. This resulted in the formation of aggregates even in solutions with a high cellobiose concentration. The previous understanding that cellobiohydrolase nucleation is inhibited by cellobiose, owing to impurity adsorption, is incorrect. The growth rate of the aggregates decreased with an increase in the cellobiose concentration, and the aggregates gradually stopped growing over time. These phenomena would be explained by an increase in chemical potential in the cellobiohydrolase monomers due to cellobiose binding. This self-inhibition mechanism has not been considered in the previous investigations of protein crystal growth in the presence of impurities; therefore, the relationships between proteins and impurities would be reexamined occasionally. Analysis of the light scattering data indicated the successful crystallization of cellobiohydrolase in the presence of cellobiose without seeds; it was gentle agitation of cellobiose-containing protein solutions during sitting-drop vapor diffusion growth. This method would be generally applicable to optimize protein crystallization protocols.
REFERENCES
- T. Bergfors, “Seeds to Crystals,” Journal of Structural Biology, Vol. 142, No. 1, 2003, pp. 66-76. http://dx.doi.org/10.1016/S1047-8477(03)00039-X
- S. M. Roberts and G. J. Davies, “The Crystallization and Structural Analysis of Cellulases (and Other Glycoside Hydrolases): Strategies and Tactics,” In: H. J. Gilbert, Ed., Methods in Enzymology, Cellulase, Vol. 510, Elsevier, 2012, pp. 141-167.
- R. Sims, M. Taylor, J. Saddler and W. Mabee, “From 1st - and 2nd -Generation Biofuels Technologies: An Overview of Current Industry and RD&D Activities,” International Energy Agency Bioenergy, OECD/IEA, Paris, 2008, p. 124.
- R. Wolfenden and M. Snider, “The Depth of Chemical and the Power of Enzymes a Catalysis,” Accounts of Chemical Research, Vol. 34, No. 12, 2001, pp. 938-945. http://dx.doi.org/10.1021/ar000058i
- R. Berner, “The Long-Term Caobon Cycle, Fossil Fuels and Atmospheric Composition,” Nature, Vol. 426, 2003, pp. 323-326.
- Y. S. Liu, Y. Zeng, Y. Luo, Q. Xu, M. E. Himmel, S. J. Smith and S. Y. Ding, “Does the Cellulose-Binding Module Move on the Cellulose Surface?” Cellulose, Vol. 16, No. 4, 2009, pp. 587-597. http://dx.doi.org/10.1007/s10570-009-9306-0
- M. Juy, A. G. Amit, P. M. Alzari, R. J. Poljak, M. Claeyssens, P. Beguin and J. P. Aubert, “3-Dimensionla Atructure of a Thermostable Bacterial Cellulase,” Nature, Vol. 357, 1992, pp. 89-91.
- Y. Hata, K. Natori, Y. Katsube, T. Ooi, M. Arai and H. Okada, “Crystallization and Preliminary-X-Ray Diffraction Studies of an Endoglucanase from Aspergillus-aculeatus,” Journal of Molecular Biology, Vol. 241, No. 2, 1994, pp. 278-280. http://dx.doi.org/10.1006/jmbi.1994.1499
- J. H. Pereira, R. Sapra, J. V. Volponi, C. L. Kozina, B. Simmons and P. D. Adams, “Structure of Endoglucanase Cel9A from the Tehrmoacidophilic Alicyclobacillus acidocaldarius,” Acta Crystallographica Section D, Vol. 65, No. 8, 2009, pp. 744-750. http://dx.doi.org/10.1107/S0907444909012773
- M. V. Liberato, W. Generoso, W. Malago Jr., F. Henrique-Silva and I. Polikarpov, “Crystallization and Preliminary X-Ray Diffraction Analysis of Endoglukanase III from Trichoderma harzianum,” Acta Crystallographica Section F, Vol. 68, No. 3, 2012, pp. 306-309. http://dx.doi.org/10.1107/S1744309112000838
- E. T. Prates, I. Stankovic, R. L. Silveira, M. V. Liberato, F. Henrique-Silva, N. Pereira Jr., I. Polikarpov and M. S. Skaf, “X-Ray Structure and Molecular Dynamic Simulations of Endoglucanase 3 from Trichoderma harzianum: Structure Organization and Substrate Recognition by Endoglucanase That Lack Cellulose Binding Module,” PLOS ONE, 2013. http://www.plosone.orgpath=10.1371/journal.pone.0059069
- T. Bergfors, J. Rouvinen, P. Lehtovaara, X. Cladentey, P. Tomme, M. Claeyssens, G. Pettersson, T. Teeri, J. Knowles and T. A. Jones, “Crystallization of the Core Protein of Cellobiohydrolase-II from Trichoderma-reesei,” Journal of Molecular Biology, Vol. 209, No. 1, 1989, pp. 167- 169. http://dx.doi.org/10.1016/0022-2836(89)90179-4
- J. Rouvinen, T. Bergfors, T. Teeri, J. K. Knowles and T. A. Jones, “Three-Dimensional Structure of Cellobiohydrolase II from Trichoderma Reesei,” Science, Vol. 249, 1990, pp. 380-386.
- A. Grassick, G. Birrane, M. Tuohy, P. Murray and T. Higgins, “Crystallization and Preliminary Crystallographic Analysis of the Catalytic Domain Cellobiohydrolase I from Talaromyces emersonii,” Acta Crystallographica Section D, Vol. 59, No. 7, 2003, pp. 1283-1284. http://dx.doi.org/10.1107/S0907444903009843
- T. Parkkinen, A. Koivula, J. Vehmaanpera and J. Rouvinen, “Preliminary X-Ray Analysis of Cellobiohydrolase Cel7B from Melanocarpus albomyces,” Acta Crystallographica Section F, Vol. 63, No. 9, 2007, pp. 754-757.
- S. Jindou, S. Petkun, L. Shimon, E. A. Bayer and R. Lamed, “Crystallization and Preliminary Diffraction Studies of CBM3b of Cellobiohydrolase 9A from Clostridium thermocellum,” Acta Crystallographica Section F, Vol. 63, No. 12, 2007, pp. 1044-1047. http://dx.doi.org/10.1107/S1744309107054644
- F. Colussi, L. C. Textor, V. Serpa, R. N. Maeda, N. Pereira and I. Polikarpov, “Purification, Crystallization and Preliminary Crystallographic Analysis of the Catalytic Domain of the Extracellular Cellulase CGHI from Trichoderma harzianum,” Acta Crystallographica Section F, Vol. 66, No. 9, 2010, pp. 1041-1044. http://dx.doi.org/10.1107/S1744309110026886
- A. Varrot, T. P. Frandsen, I. von Ossowski, V. Boyer, S. Cottaz, H. Driguez, M. Schulein and G. J. Davis, “Structural Basis for Ligand Binding and Processivity in Cellobiohydrolase Cel6A from Humicola insolens,” Structure, Vol. 11, No. 7, 2003, pp. 855-864. http://dx.doi.org/10.1016/S0969-2126(03)00124-2
- K. Eckert, A. Vigouroux, L. Lo Leggio and S. Morera, “Crystal Structures of A. acidocaldarius Endogulucanase Cel9A Complex with Cello-Oligosaccharides,” Journal of Molecular Biology, Vol. 394, No. 1, 2009, pp. 61-70. http://dx.doi.org/10.1016/j.jmb.2009.08.060
- W. Ubhayasekera, I. G. Munoz, A. Vasella, J. Stahlberg and S. L. Mowbray, “Structure of Phanerochaete chrysosporium Cel7D in Complex with Products and Inhibitors,” FEBS Journal, Vol. 272, No. 8, 2005, pp. 1952-1964. http://dx.doi.org/10.1111/j.1742-4658.2005.04625.x
- B. R. Thomas, P. G. Vekilov and F. Rosenberger, “Effects of Microheterongeneity in Hen-Egg White Lysozyme Crystallization,” Acta Crystallographica Section D, Vol. 54, No. 2, 1998, pp. 226-236. http://dx.doi.org/10.1107/S0907444997010676
- W. Eberstein, Y. Georgalis and W. Saenger, “Molecular Interactions in Crystalling Lysozyme Solutions Studied by Photon Correlation Spectroscopy,” Journal of Crystal Growth, Vol. 143, No. 1-2, 1994, pp. 71-78. http://dx.doi.org/10.1016/0022-0248(94)90369-7
- S. Tanaka, M. Ataka, K. Onuma and T. Kubota, “Rationalization of Membrane Protein Crystallization with Polyehtylene Glycol Using a Simple Depletion Model,” Biophysical Journal, Vol. 84, No. 5, 2003, pp. 3299-3306. http://dx.doi.org/10.1016/S0006-3495(03)70054-X
- K. Onuma and N. Kanzaki, “Size Distribution and Intermolecular Interaction of Laminin-1 in Physiological Solution,” Journal of Physical Chemistry B, Vol. 107, No. 42, 2003, pp. 11799-11804. http://dx.doi.org/10.1021/jp0355298
- D. L. Zechel, A. B. Boraston, T. M. Gloster, C. M. Boraston, J. M. Macdonald, D. Matthew, G. Tilbrook, R. V. Stick and G. J. Davis, “Iminosugar Glycosidase Inhibitors: Structural and Thermodynamic Dissection of the Binding of Isofagomine and -Glucosidases 1-Deoxynojirimycin to Two,” Journal of American Chemical Society, Vol. 125, No. 47, 2003, pp. 14313-14323. http://dx.doi.org/10.1021/ja036833h
- N. Kanzaki, T. Ueda and K. Onuma, “Intermolecular Interaction of Actin Revealed by Dynamic Light Scattering Technique,” Journal of Physical Chemistry B, Vol. 110, No. 6, 2006, pp. 2881-2887. http://dx.doi.org/10.1021/jp054865g
- S. W. Provencher, “A Constrained Regularization Method for Inverting Data Represented by Linear Agebraic or Integral Equations,” Computer Physics Communications, Vol. 27, No. 3, 1982, pp. 213-227. http://dx.doi.org/10.1016/0010-4655(82)90173-4
- S. E. Ingles, A. Katzenstein, W. Schlenker and K. Huber, “Time-Resolved Recording of Ionic Dyestuff Aggregation by Static Light Scattering,” Langmuir, Vol. 16, No. 7, 2000, pp. 3010-3018. http://dx.doi.org/10.1021/la9903649
- K. Onuma, A. Oyane, K. Tsutsui, K. Tanaka, G. Treboux, N. Kanzaki and A. Ito, “Precipitation Kinetics of Hydroxyapatite Revealed by Continuous-Angle Laser Light Scattering Technique,” Journal of Physical Chemistry B, Vol. 104, No. 45, 2000, pp. 10563-10568. http://dx.doi.org/10.1021/jp002697g
- T. Witte, B. Decker, J. Mattay and K. Huber, “Formation of Branched Callxarene Aggregates-A Time-Resolved Static Scattering Study,” Journal of American Chemical Society, Vol. 126, No. 30, 2004, pp. 9276-9282. http://dx.doi.org/10.1021/ja0493291
- K. Onuma, A. Watanabe, N. Kanzaki and T. Kubota, “Association Kinetics of Wild-And Mutant-Type Ynd1p in Relation to Quality of Grown Crystals,” Journal of Physical Chemistry B, Vol. 110, No. 49, 2006, pp. 24876- 24883. http://dx.doi.org/10.1021/jp0643146
- K. Onuma, N. Furubayashi, F. Shibata, Y. Kobayashi, S. Kaito, Y. Ohnishi and K. Inaka, “Anomalous Effect of Poly(ethylene)glycol on Intermolecular Interaction and Protein Association,” Crystal Growth & Design, Vol. 9, No. 5, 2009, pp. 2517-2524. http://dx.doi.org/10.1021/cg900019e
- P. N. Pusey and R. J. A. Tough, “Particle Interactions,” In: R. Pecora, Ed., Dynamic Light Scattering, Plenum, New York, 1985, pp. 85-179. http://dx.doi.org/10.1007/978-1-4613-2389-1_4
- D. N. Petsev and P. G. Vekilov, “Evidence of Non-DLVO Hydration Interactions in Solutions of the Protein Apoferritin,” Physical Review Letters, Vol. 84, No. 6, 2000, pp. 1339-1342. http://dx.doi.org/10.1103/PhysRevLett.84.1339
- J. Narayanan and X. Y. Liu, “Protein Interactions in Undersaturated and Supersaturated Solutions: A Study Using Light and X-Ray Scattering,” Biophysical Journal, Vol. 84, No. 1, 2003, pp. 523-532. http://dx.doi.org/10.1016/S0006-3495(03)74871-1
- K. C. Holmes, W. Gebhard, D. Popp and W. Kabsch, “Atomic Model of the Actin Filament,” Nature, Vol. 347, 1990, pp. 44-49.
- X. Chen, R. K. Cook and P. A. Rubenstein, “Yeast Actin with a Mutation in the “Hydrophobic Plug” between Subdomains 3 and 4 (L266D) Displays a Cold-Sensitive Polymerization,” Journal of Cell Biology, Vol. 123, 1993, pp. 1185-1195. http://dx.doi.org/10.1083/jcb.123.5.1185
NOTES
*Corresponding author.