1. Introduction
Some number of bi-center clusters of the T2Sin type were primarily studied for the transition metals (T = Cr, Mn) [1] and then for the transition metals T = Fe, Co, Ni [2] , with a number n in the range 1 £ n £ 8 by the density functional theory (DFT) in the PBE approach. The geometry of small Fe2Sin is characterized by large number of local minima what prevents the predictable change of different spin states with the increasing of excitation energy of a system. The property of Fe2 molecule inside a large siliceous cluster seems to be more controlled in the case of its “good packing” into the cluster. The later can be achieved in the case of Fe2Si18 cluster, which electronic properties are investigated below in a framework of multireference configuration interaction approach with the singlet and double replacements into the external space. The DFT methods are not suitable enough for obtaining the correct sequence of exited states and especially for the description of a singlet state in the systems containing the transition elements, the Fe-atom especially, see in this connection the table 6 in [3] where some number of DFT methods are compared with configuration interaction (CI) approach for the case of FeSi4 cluster.
The search of the two-state systems represents intriguing story in the modern investigations of a quantum computer systems. We mention here the point-like NV defect state in diamond [4] [5] and molecular-like pair of defect spin states (VSi-VC) in carbide [6] and the search of the two-level states in biophysical molecular structures [7] [8] . The singlet state of Fe2Si18 cluster would have a special interest in this connection.
2. Calculation Procedure
The accuracy of multiconfigurational method for the Si subsystem can be demonstrated for Si2 molecule with the ground state configuration 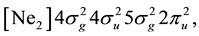 term
term , where [Ne2] means the closed shell of the neon dimer in the Hartree-Fock approximation. The competing
, where [Ne2] means the closed shell of the neon dimer in the Hartree-Fock approximation. The competing 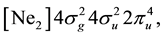
 configurations generate the ground state terms
configurations generate the ground state terms 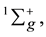 3Õu. The calculations were performed using the GAMESS (2011, 2013) program [9] in the atomic aug-cc-pVTZ basis set. The construction of molecular orbitals (МОs) for the second-order CI (SOCI) problem is carried out by the complete active space self-consistent field (CASSCF) method. The [5sg 2pu]4 active space was used with averaging over two terms with the weights: 1/3 for the
3Õu. The calculations were performed using the GAMESS (2011, 2013) program [9] in the atomic aug-cc-pVTZ basis set. The construction of molecular orbitals (МОs) for the second-order CI (SOCI) problem is carried out by the complete active space self-consistent field (CASSCF) method. The [5sg 2pu]4 active space was used with averaging over two terms with the weights: 1/3 for the  state and 2/3 for 3Õu. Based on the obtained canonized MOs we considered the SOCI type excitations from the reference space [4sg 4su 5sg 2pu]8 for the whole virtual space. After optimization of bond Si?Si length in accordance with the spectroscopic data [10] it is found that the term of the Si2 ground electronic state is
state and 2/3 for 3Õu. Based on the obtained canonized MOs we considered the SOCI type excitations from the reference space [4sg 4su 5sg 2pu]8 for the whole virtual space. After optimization of bond Si?Si length in accordance with the spectroscopic data [10] it is found that the term of the Si2 ground electronic state is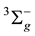 , the equilibrium bond length is r(Si-Si) = 2.245Å, which is consistent with its experimental value r(Si-Si) = 2.246 Å. The first excited state turns out to be 3Õu with the vertical excitation energy of 0.273 eV at a bond length of the ground state.
, the equilibrium bond length is r(Si-Si) = 2.245Å, which is consistent with its experimental value r(Si-Si) = 2.246 Å. The first excited state turns out to be 3Õu with the vertical excitation energy of 0.273 eV at a bond length of the ground state.
For the subsequent calculations of the Fe2Si18 clusters a special basis set was constructed for the iron atom. The core was treated with the 6-31G(f) basis set to which three additional s, p, and d type Gaussian functions were then added. The exponents of the added functions were optimized with retaining all other functions of the 6-31G(f) basis set. Various methods (ROMP2, MCQDPT [11] , and the coupled cluster method in the CR-CC(2,3) variant [12] [13] yield close values of the exponents in the optimization of the total energy for the ground state of the iron atom (term 5Dg); in the CR-CC(2,3) method all indices proved to be z(s) = 0.280829, z(p) = 0.120767, z(d) = 0.136335.
The calculation scheme looks as following. The total spin S of the Fe2 molecule is equal to S = 4 in the ground state and it can induce some spin polarization effect on the surrounding Si atoms. We performed first UHF calculations to take into account this possibility into the consideration with a sufficiently large value of a spin projection MS = 10 that suppose the presence of 8 unpaired electrons in 3d-shells of both Fe-atoms and 12 ones of spin-polarized Si atoms. The obtained UHF natural orbitals were used as the starting orbitals for the next CASSCF calculation with a single high-spin configuration with S = 10 including 20 singly occupied molecular orbitals (MOs) as active orbitals. The obtained CASSCF MOs were transformed to the canonic form with the use of the GAMESS algorithm procedure. These canonized active MOs were divided into three subsets: 6 orbitals with the lowest orbital energies, 8 orbitals resembling 3d-shell states of iron atoms and 6 orbitals with the highest orbital energies. After that three subsets were used to construct three orbital subspaces of the restricted active space self-consistent field (RASSCF) method. The maximum electronic excitation levels between subspaces were allowed to be 2, for the details see also [3] . There were imposed no restrictions on the symmetry of the cluster during the geometry optimization. The obtained electronic properties are summarized in the Table 1 for the total spin S = 4 (ground state) and for the first singlet state, other spin states S = 3, 2, 1 are described in the supplementary information, Table A. All molecular structures were depicted by means of MacMolPlt program [14] .
The most laborious part in the calculations is of course the investigation of electronic properties in the first singlet state, its description takes Q = 22065484 determinant functions compared to Q = 295795 determinant functions for the ground S = 4 state. Electronic properties are found to be well understandable, the 8 once occupied MOs are well localized on the two Fe atoms, hence all spin states with S = 4, 3, 2, 1, 0 represent the same electronic configuration. Some number of natural orbitals (NOs) with the occupation numbers n = 2, 1, 0 are represented in the Table 2(a) for the ground state and in the Table 2(b) for the singlet state.
3. Conclusions
The most important data are summarized below in the Table 3.
![]()
Table 1. Electronic structure of Fe2Si18 cluster in the ground S = 4 and in the excited singlet S = 0 spin states. Distances in Å, dipole moment in D-units.
(a) ![]() (b)
(b) ![]()
Table 2. (a) Some natural-orbitals for different occupation numbers n in the ground state (S = 4) of Fe2Si18. (b) Some natural-orbitals for different occupation numbers n in the singlet state (S = 0) of Fe2Si18.
![]()
Table 3. Dipole moments (d) in Debye u., Mulliken charge difference Δq for the first and second Fe atoms, transition energy ΔE (eV) for the different spin states of Fe2Si18 cluster.
We conclude: 1) the excitation energy between neighboring spin states is a decreasing function of the transition energy, 2) the charge difference Δq is nearly stable, 3) the total dipole moment is a drastically decreasing function of the transition energy ΔΕ. The last observation is most importance and it can be understood as following. The large positive charge on one Fe-atom, it denoted by number 1 in the Table 1, induces relative large negative charges on the neighboring Si atoms, as it represented in the Table 1. The second Fe-atom has a noticeable less charge and induces smaller negative charges on the surrounding Si-atoms. The resulting dipole moment, which is independent on the method of the charge definition, becomes relative small. The dipole moment in the singlet state is so small and it can be taken practically as zero considering inescapable uncertainties in the calculations. The sequence of the spin states is the same as for the Fe2 molecule in the DFT (B3P86) approach [15] , but the tendency in the energy level intervals is quite different, we have the decreasing level spacing with the increase of the energy that corresponds to free Fe-atom data.
The symmetric charge distribution is unstable, there arise some kind of “up and down states” in the sense of the charge distribution in the degenerate electronic state. This conclusion can be checked by the measurement of Mössbauer spectra on the two Fe-atoms. Only the stable electronic structures were described above, the problem of their transformations is to be discussed separately.
Supplementary Information
![]()
Table A. Electronic properties of Fe2Si18 cluster in the spin S = 3, 2, 1 states. The total energy E (a.u.), the excitation energy ΔΕ (eV), dipole moments d (D), equilibrium distances (Å) (numbering of atoms is the same as in the Table 1).
NOTES
*Corresponding author.