1. Introduction
Nicotine {(S)-3-(1-Methyl-2-pyrrolidinyl)pyridine} is a weakly basic bicyclic tertiary amine mostly found in tobacco plants (up to 2% - 8%) as well as in few vegetables such as potatoes and tomatoes in minor quantities. There has been tremendous interest in nicotine chemistry, related pharmacology and metabolism primarily due to widespread exposure of nicotine among people through recreational use of tobacco products [1]. Being a molecule with one chiral centre, nicotine can exist in two sterioisomeric forms viz. (R) & (S) respectively (Figure 1), with latter being the predominant enantiomer found in tobacco plants (>99%) [2] and is known to exhibit significant pharmacologic activity.
Though nicotine is traditionally known for its dependence potential, lately nicotine has attracted much attention because of its pharmacological effects on central nervous system (CNS) diseases. In particular, (S)-nicotine may have beneficial effects in the treatment of Parkinson’s disease (PD), Alzheimer’s disease (AD), Tourette’s syndrome, schizophrenia, attention-deficit hyperactivity disorder (ADHD), smoking cessation, epilepsy, and depression [1]. Nicotine is administered in low dose levels to patients seeking relief from withdrawal symptoms associated with cessation of tobacco consumption. Products in this category, known as nicotine replacement therapy, include nicotine gums, nicotine lozenges, nicotine pouches, nicotine patches, nicotine nasal spray, mouth spray etc. In all these products, nicotine derived from tobacco is used as active pharmaceutical ingredient (API) either in its free form or as salt/resinate. However, nicotine derived from tobacco extraction is typically contaminated with a few inseparable alkaloids known as nicotine related substances. These alkaloids include nornicotine, anabasine, anatabine, myosmine, nicotyrine and cotinine. United States Pharmacopeia regulates certain levels for these impurities to ascertain nicotine purity. Additionally products utilizing nicotine sourced from tobacco falls under the regulatory arms of government bodies (For e.g. US FDAs Tobacco Control Act definition of “tobacco product” includes any product made or derived from tobacco and intended for human consumption, including any component, part or accessory of a tobacco product). Until recently, synthetic nicotine wasn’t considered with-in US FDAs purview for regulation. These factors combined have prompted researchers to develop an efficient route to enantiomerically pure nicotine as it will be devoid of related impurities. Unlike tobacco derived nicotine, synthetic nicotine is colorless, odorless and does not impart harsh taste.
A thorough literature survey of earlier nicotine synthetic methods was conducted. The first synthesis of optically active nicotine was achieved by Chavdarian and co-workers in 1982 [3]. Other reported methods include strategies that utilize β-allyldiisopinocampheylborane to form chiral alcohol [4] [5] [6]. Another literature example revealed synthesis of nornicotine enantiomers by treating racemic nornicotine with optically pure (-) menthyl chloroformate, separating the N’-(menthoxycarbonyl) nor nicotine diastereomers and subsequent hydrolysis of the separated diastereomers to yield optically pure nornicotine [7]. Carolin
![]()
Figure 1. Structures of (R) and (S) nicotine.
Welter and co-workers reported enantioselective synthesis of (R) and (S)-nicotine based on Ir-catalysed allylic amination [8]. Other approaches to synthesis of nicotine were also reported in [9] - [14].
Synthesis of nicotine has been patented by few companies. Examples can be seen in synthesis and resolution of nicotine patented by NJOY, LLC (US) [15]. London based Zanoprima Life Sciences Ltd, have a patented method to produce (S) nicotine utilizing enzymatic reduction of myosmine as a key step [16]. United States based Next Generation Labs, LLC have patented their technology for the preparation of (R, S) nicotine [17]. Synthesis of nicotine was also patented by few other organizations such as CSIR India [18], Siegfried and Contraf-Nicotex-Tobacco GmbH [19] [20].
The strategy employed in this work is novel and differntiative from earlier reported synthesis in that we have successfully demonstrated use of (S)-Ibuprofen as a chiral resolving agent leading to the synthesis of (R) and (S) nicotine, circumventing any late stage separation of isomers. Also (S)-Ibuprofen can be recovered during the process (Step 4, Scheme A) and can be reused, which will make the process cost effective.
2. Synthesis of (R) and (S) Nicotine
Synthesis of (R) and (S) nicotine outlined in scheme A (separation and isolation of racemic alcohol in to enantiomerically pure forms) and Scheme B (conversion of separated enantiomerically pure alcohols in to (R) and (S) nicotine respectively).
2.1. Scheme A
Synthesis and separation of (R)and (S)1-(pyridin-3-yl)but-3-en-1-ol (R)-6a and (S)-6a starting from (S)-Ibuprofen (3)
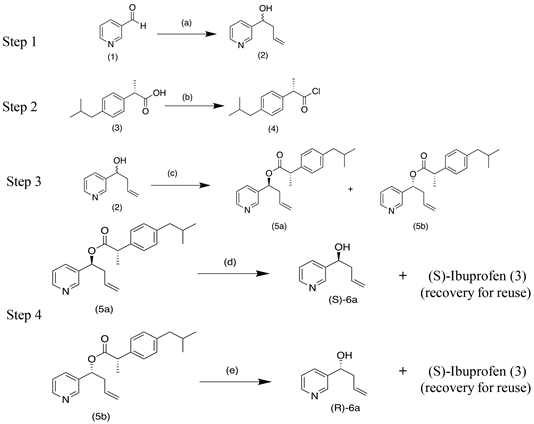
Conditions: (a) Allyl bromide, Tin, water diethyl ether, aq HBr, RT, 3 hrs (b) SOCl2, Benzene, Reflux 3 hrs (c) (S)-Ibuprofen acid chloride(4), Pyridine, DCM, 0˚C-RT, 6 hrs (d) & (e) 1N NaOH, MeOH, RT, 2 hrs.
Experimental Pertaining to Scheme A
(a) Synthesis of (R,S)-1-(pyridin-3-yl)but-3-en-1-ol (2)
Tin metal powder (1.8 g, 15.16 mmol; Sd Fine Chem Ltd, 99.9%) was taken in a dry 100 ml single neck RB flask. A 1:1 mixture of water and diethyl ether (20ml each) was added to the flask and was stirred under magnetic bar which was followed by the addition of allyl bromide (2.2 g, 18.18 mmol; Sd Fine Chem Ltd, 98%). The mixture was stirred for 5 mins before adding aqueous HBr (1 ml; Sd Fine Chem Ltd, 48.0%) with stirring. Pyridine-3-carbaldehyde (1) (1.07 g, 9.98 mmol; Sigma Aldrich, 98%) was added to the mixture and the resulting mixture was stirred for 3 hrs at RT as monitored by TLC. Work up was done by basifying the medium to pH of 7.5 using 10% aqueous sodium bicarbonate (Sd Fine Chem Ltd, 99.7%). Crude mixture was transferred to a separating funnel and extracted using dichloromethane (30 ml × 3 times). The combined organic layer was dried over anhydrous sodium sulfate and concentrated on a rotary evaporator to afford racemic alcohol (2) as pale yellow oil upon distillation of crude mass. (1.39 g, 94% yield).
Characterization: IR (neat): 3215.45 cm−1; 1641.39 cm−1, 1579.58 cm−1
(b) Synthesis of (S)-Ibuprofen acid chloride (4)
(S)-Ibuprofen (4.8 g, 23.27 mmol; Strides Pharma science Ltd, > 99%) was taken in a dry 100 ml three neck RB flask dissolved in benzene (30 ml) under N2 atmosphere. Added thionyl chloride (3.46 g, 29.08 mmol; Sd Fine Chem Ltd, 99.5%) and the mixture was heated to reflux for 3 hrs, post which excess thionyl chloride and benzene were removed through distilling the reaction mass under vacuum. Resulting (S)-Ibuprofen acid chloride (4) was taken for next step immediately.
(c) Synthesis and separation of diastereomeric esters (S)-1-(pyridine-3-yl)but-3-en-1-yl(S)-2-(4-isobutylphenyl) propanoate (5a) and (R)-1-(pyridine-3-yl) but-3-en-1-yl(S)-2-(4-isobutylphenyl) propanoate (5b)
(S)-Ibuprofen acid chloride (4) (5.2 g, 23.13 mmol) was cooled in dry dichloromethane (50 ml) to 0˚C. To this was added racemic alcohol (2) (2.9 g, 19.43 mmol) previously dissolved in dichloromethane dropwise. Finally pyridine (2 ml) was added to the mixture and reaction was continued at RT for about 6 hrs. Crude reaction mixture was washed with water and extracted using dichloromethane (30 ml × 3 times). The combined organic layer was dried over anhydrous sodium sulfate and concentrated on a rotary evaporator to yield crude mass, taken for column chromatographic separation. The formed diastereomers were separated carefully using column chromatography. Conditions: Silica gel (100 - 200 mesh), Eluent (Gradient elution 0% - 10% ethyl acetate in hexane). Special care was taken to isolate both the fractions in order to attain good purity (4.22 g, 54% yield including both diastereomers).
Characterizations:
Compound (5a) - IR (neat): 1734.81 cm−1; 1642.26 cm−1, 1598.10 cm−1
GC-MS: [M+] at 337.24, [M+-CH3] at 322.22
Compound (5b) - IR (neat): 1734.99 cm−1; 1643.60 cm−1, 1578.17 cm−1
GC-MS: [M+] at 337.22, [M+-CH3] at 322.24
(d) Synthesis of (S)-1-(pyridin-3-yl) but-3-en-1-ol (S)-6a
1N NaOH (10 ml; Sd Fine Chem Ltd, 98%) was added dropwise to an ice cooled solution of ester (5a) (3.2 g, 9.5 mmol) in methanol (10 ml). Solution was stirred at RT for 2 hrs as monitored by (TLC). Reaction mixture was concentrated on a rotary evaporator and resulting crude mass was washed with water and extracted using dichloromethane (30 ml × 2 times). The combined organic layer was dried over anhydrous sodium sulfate and concentrated to yield optically active homoallylic alcohol (S)-6a (1.1 g, 78% yield).
Characterization: IR (neat): 3350.33 cm−1; 1641.51 cm−1; 1595.69 cm−1; 1581.69 cm−1
(e) Synthesis of (R)-1-(pyridin-3-yl) but-3-en-1-ol (R)-6a
1N NaOH (6 ml; Sd Fine Chem Ltd, 99.5%) was added dropwise to an ice cooled solution of ester (5b) (1.7 g, 5.04 mmol) in methanol (6 ml). Solution was stirred at RT for 2 hrs as monitored by (TLC). Reaction mixture was concentrated on a rotary evaporator and resulting crude mass was washed with water and extracted using dichloromethane (30 ml × 2 times). The combined organic layer was dried over anhydrous sodium sulfate and concentrated to yield optically active homoallylic alcohol (R)-6a (0.5 g, 67% yield).
Characterization: IR (neat): 3206.41 cm−1; 1640 cm−1; 1593 cm−1; 1579.39 cm−1
Data on specific rotation was recorded for the alcohols (S)-6a and (R)-6a and was compared against that reported in literature [21]. Same is captured in Table 1 below.
![]()
Table 1. Data on specific rotation for separated alcohols; (S)-6a and (R)-6a.
2.2. Scheme B
Synthesis of (R)-nicotine (10) from (S) 1-(pyridin-3-yl) but-3-en-1-ol (S)-6a
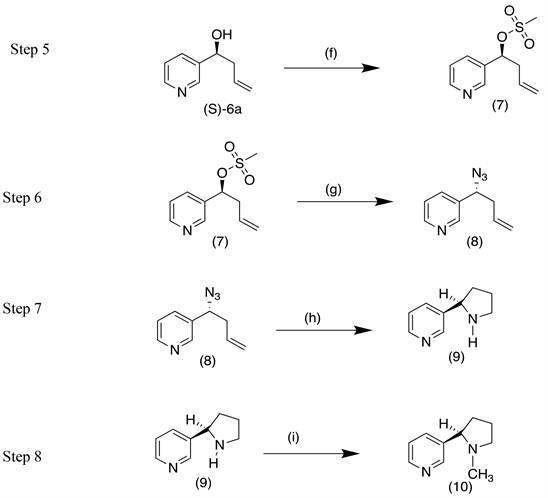
Conditions: (f) CH3SO2Cl, Et3N, 0˚C, 1 hr (g) NaN3, DMF, 60˚C, 4 hr (h) BH3-THF (1M), Cyclohexene, THF, (0˚C to −15˚C), RT 12 hr (i) HCOOH, HCHO reflux 9 hrs.
Experimentals Pertaining to Scheme B
(f) Synthesis of (S)-1-(pyridin-3-yl) but-3-en-1-yl-methanesulfonate (7)
Triethylamine (0.726 g, 7.17 mmol; Sigma Aldrich, 99.5%) was added to an ice cooled solution of (S) 1-(pyridin-3-yl) but-3-en-1-ol (S)-6a (0.5 g, 3.35 mmol) taken in dry dichloromethane (20 ml). Mixture was stirred for 5 mins, followed by dropwise addition of methane sulfonyl chloride (0.65 g, 5.67 mmol; Sd Fine Chem Ltd, 98%). Reaction mixture was stirred for 1 hr at RT as monitored by TLC. Post consumption of starting material, reaction mixture was diluted with water (20 ml) and layers were separated. Aqueous layer was further extracted with dichloromethane (20 ml × 3 times). The combined organic layer was dried over anhydrous sodium sulfate and concentrated to yield (S)-1-(pyridin-3-yl) but-3-en-1-yl-methanesulfonate (7) (0.7 g, 92% yield) which was taken immediately for next step.
(g) Synthesis of (R)-3-(1-azidobut-3-en-1-yl) pyridine (8)
Sodium azide (0.3 g, 4.61 mmol; Sd Fine Chem Ltd, 99.5%) was added in one portion to a solution of (S)-1-(pyridin-3-yl) but-3-en-1-yl-methanesulfonate (7) (0.7 g, 3.07 mmol) taken in dry DMF (10 ml). Resulting mixture was stirred at 60˚C for 4 hrs. Reaction mixture was diluted with ether (15 ml) and layers separated. Aqueous layer was further extracted with diethyl ether (15 ml × 3 times). The combined organic layer was dried over anhydrous sodium sulfate and concentrated to yield (R)-3-(1-azidobut-3-en-1-yl) pyridine (8) as crude mass which was purified by column chromatography. (0.5 g, 93.4% yield).
Characterization: IR neat: 2092.74 cm−1, 1676.46 cm−1, 1643.17 cm−1, 1591.15 cm−1
(h) Synthesis of (R)-nornicotine(9)
To a magnetically stirred solution of freshly distilled cyclohexene (1.8 g, 21.95 mmol) in dry THF was added BH3-THF solution (1M, 11 ml; Sigma Aldrich) dropwise. Resulting white suspension was stirred for 2 hrs at 0˚C. To this was added (R)-3-(1-azidobut-3-en-1-yl) pyridine (8) (0.647 g, 3.72 mmol) dissolved in THF (20 ml) at −15˚C. Reaction was continued to stir at RT for 12 hrs. Reaction mixture was quenched with methanol. This was followed by diluting the mixture with diethyl ether (20 ml) and was extracted with 0.1N HCl (10 ml × 2 times). The combined aqueous layer was treated with 30% aq NaOH solution until pH 13 - 14, saturated with NaCl and extracted with diethyl ether (20 ml × 2 times). The combined organic layer was dried over anhydrous sodium sulfate and concentrated to yield (R)-nornicotine (9) (0.35 g, 64% yield).
Characterization: IR neat: 3323.17 cm−1, 2930.36 cm−1, 2856.58 cm−1
(i) Synthesis of (R)-nicotine(10)
Formaldehyde (0.42 ml; Sd Fine Chem Ltd., 37% - 41% w/v) and formic acid (0.48 ml; Sd Fine Chem Ltd., 98%) were added to (R)-nornicotine (0.3 g, 2.02 mmol) (9). Reaction mixture was heated to reflux for 9 hrs as monitored by TLC. Reaction mixture was diluted with diethyl ether (15 ml) and water (15 ml), layers were separated and aqueous layer was saturated with NaCl and extracted using diethyl ether. The combined organic layer was dried over anhydrous sodium sulfate and concentrated to yield (R)-nicotine as crude, which was distilled to get pure (R)-nicotine (10) (0.2 g, 61% yield, HPLC purity-99.4%, Chiral purity 80.7%).
2.3. Synthesis of (S)-nicotine from (R) 1-(pyridin-3-yl) but-3-en-1-ol (R)-6a
Synthesis of (S)-nicotine was achieved using chiral alcohol (R)-6a employing similar conversions and reaction conditions as employed for synthesis of (R)-nicotine as shown in Scheme B.
3. Separation and Quantification of (R) and (S) Nicotine through Chiral HPLC
Many methods are available for the separation and quantification of (R) and (S) nicotine in racemic mixture [22] - [27]. Based on these available literature methods, a simple one-step chiral high performance liquid chromatography (HPLC) method was developed to separate (R) and (S) enantiomers of nicotine using chiral amylose column.
(S)-nicotine and (R)-nicotine standards were procured from Sigma Aldrich for the quantification of synthesized (R) and (S) nicotine. The purity of these standards was determined using HPLC. Purity of (S)-nicotine standard was 100%, whereas purity of (R)–nicotine was observed as 97.5%.
(R) and (S)-nicotine in corresponding samples were determined by extracting the sample with n-hexane. The extracted solution was analyzed using HPLC equipped with Diode Array Detector (DAD) at 254 nm by using amylose column (5 µm × 4.6 mm × 250 mm), Flow was maintained at 0.5 mL/min, oven temperature was at 30˚C, injection volume was 10 µl and mobile phase was n-hexane and isopropanol in 98:2 ratio in isocratic condition. Total runtime was 30 min.
The peak resolution of (S)-nicotine and (R)-nicotine is found to be good (Figure 2). The developed method has good precision and recovery rates for (S)-nicotine and (R)-nicotine (i.e., 97.2% & 98.9%, respectively.) Table 2 summarizes chiral purity obtained for synthesized (R) and (S) Nicotine using developed method.
![]()
Table 2. Chiral purity results by HPLC for synthesized(R) and (S)-nicotine.
![]()
Figure 2. Chromatogram of (R) and (S)-nicotine.
4. Conclusion
Synthesis of (R) and (S) nicotine was achieved via a novel approach, which effectively separated racemic homoallylic alcohol intermediate (2) to its optically pure forms (S)-6a & (R)-6a. In this work, we reported the use of (S)-Ibuprofen as a chiral resolving agent, which facilitated the separation of racemic alcohol (2) through formation of separable Ibuprofen ester diastereomers and subsequent hydrolysis. Separation of racemic alcohol as reported here was found to be simple and straightforward method without requiring the need of expensive catalysts or enzymes.
Acknowledgements
The authors would like to thank ITC Limited, ITC Life Sciences and Technology Centre, Peenya Industrial Area, Phase I, Bangalore, India for providing necessary infrastructure to carry out experiments and analysis.
Authors would also like to thank Strides Pharma Science Ltd., Bangalore, India for arranging (S)-Ibuprofen (as a gift sample), a key chiral resolving agent utilized for the synthesis and separation of racemic homoallylic alcohol intermediate.