An Aminopyrrolidinyl Phosphonates—A New Class of Antibiotics: Facile Synthesis and Predicted Biological Activity ()
1. Introduction
Natural antibiotics are produced by many microorganisms and are of great interest in medicine and pharmacology [1] [2] [3] [4]. It is well known that some antibiotics, such as K-26, SF-2513, FR-33289, and SF-2312 contain a C-P (carbon-phosphorus) bond that belongs to the category of phosphonates [5] [6] [7] [8]. The targeted synthesis of phosphonates for pharmacological utility is also fairly well described in several reviews [9] [10] [11] [12] [13].
For instance, a pyrrolidinyl phosphonates class (known as antibiotic SF-2312) is a natural antibiotic which is produced by the actinomycete Micromonospora sp. It has not only showed activity as an enolase inhibitor but also found to be one of the most potent natural inhibitors of glycolysis which in turn inhibit cell proliferation [14] [15] [16] [17] [18]. Later, the synthesis of this antibiotic and its analogues has been described by various researchers [19] [20] [21].
In the same context, pharmacologists always agree that the biological activity of both natural and synthetic compounds is related to the chemical structure [22]. Therefore, there are a large number of available computer programs that can evaluate the probability of an organic compound activity to be a drug [23] [24]. For instance, PASS computer program contains a library with information of about 1 million chemical compounds and more than 10,000 biological activities [25]. An algorithm for the practical use of PASS has been described in detail in several publications [26] [27] [28].
Previously, few pyrrolidinylphosphonates and pyrrolidinylphosphonic acids (1-12, Figure 1) were synthesized for the purpose of testing their biological activity. Generally, they were obtained either from readily available pyrrolidine ring or by multistep cyclization reactions. But there is no one general direct method to produce them and a brief summary for their synthesis is described below:
Initially, the antibiotic SF-2312 (1) and its analogue (3) were synthesized by a multistep reaction sequence in which ethyl diethoxyphosphorylacetate was converted to N-benzyloxy-2-(diethoxyphosphoryl)-pent-4-enamide followed by oxidative cleavage and hydrolysis [21]. Later, studies showed that their antibiotic activity is due to the inhibition of a glycolytic enzyme called enolase [14] [15] [16]. The reaction of pentanedial with acetamide and acetyl chloride in the presence of phosphorylating agent afforded the pyrrolidinyldiphosphonic acid (2) [29]. The combination of the Kabachnik–Fields reaction with a subsequent ring closure of 5-chloro-2-pentanone with ammonia and diethyl phosphonate produced (4) [30].
The pyrrolidinylphosphonates (5-6) were prepared from readily available 2-methylpyrroline and diethyl phosphite [31]. The SF-2312 (1) antibiotic analogues (7-10) were obtained using 1-benzyloxy-3-bromopyrrolidin-2,5-dione through Michaelis-Arbusov reaction with trialkyl phosphites followed by alkylation. However, their biological activity was not determined [19] [20].
Asymmetric synthesis of β-amidophosphonate (11) was achieved by Diels–Alder reaction between the vinylphosphonate and chiral aminodiene [32]. Finally, three-component decarboxylative coupling of proline with aldehydes and dialkyl phosphite was used to afford the corresponding pyrrolidinylphosphonates (12) [33].
2. Results and Discussion
We recently reported the synthesis of N-substituted pyrrolidinyl-methylphosphonate (14) by addition of amines to (Z)-diethyl (5-chloropent-1-en-1-yl)phosphonate 13 (Scheme 1) [34].
![]()
Figure 1. Pyrrolidinyl phosphonates 1-12.
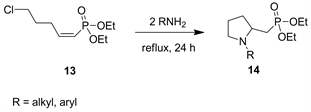
Scheme 1. Formation of pyrrolidines N-substituted pyrrolidinyl-methylphosphonate 14.
In addition, we investigated the reaction of 4-chloro-1-butentynylphosphonate 15 with an equivalent number of amines which gave 2-amino-cyclobutenylphosphonates [35]. Interestingly, a different novel class of compounds (17a-h) was obtained by tuning of the reaction conditions.
Being encouraged by these results, we determined to study amine addition on a shorter chain alkynylphosphonate that has not been explored before. Accordingly, we prepared 4-chloro-1-butentynylphosphonate 15 in our lab by substituting the hydroxy group in but-3-yne-1-ol using thionyl chloride under reflux. After isolation of the product by distillation, it was lithiated using n-BuLi, and was reacted with chlorophosphonates.
Herein, we report a very facile method for the synthesis of novel aminopyrrolidinyl phosphonates (17a-h) and their predicted biological activity using the PASS program.
Thus, when three equivalents of i-propylamine were added to (15), the pyrrolidine structure diethyl (1-isopropyl-3-(isopropylamino)pyrrolidin-2-yl)phosphonate (17a) was gained in 80% yield (Scheme 2). Similarly, products (17b-h) were produced in (74% - 79%) isolated yield by treatment of 15 with a different primary amine.
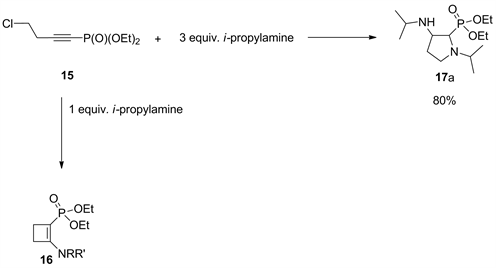
Scheme 2. Formation of pyrrolidinylphosphonate 17a.
The reaction was carried out at 25˚C for 12 h. and the products 17 were isolated by silica gel column chromatography in good yields (74% - 80%) and were characterized by NMR, GC/MS, and by elemental analysis. The multiplets in the regions ~(1.3 - 2. 0 ppm), ~(2.1 - 3.0 ppm) in the 1H NMR spectrum, precisely the doublet of doublet in the region ~2 ppm that corresponds to the hydrogen on C1 split by phosphorus and the hydrogen on C2 which resonated as a multiplet at ~3 ppm, together with the carbons in the regions ~(30, 32, 45, 55 ppm) in the 13C NMR spectra are indicative of the pyrrolidine ring. Besides, the 31P NMR that resonated chemical shifts at ~31 ppm, the GC/MS and the elemental analysis are all evidence of structure 17. These results were also supported by 2-D Cosy and 1H-13C HSQCSI NMR of 17d in which a high correlation was observed.
As described above, pyrrolidines 1-12 are of predicted biologically active compounds despite the multistep procedures for their preparation.
This process represents a general one-pot method for the synthesis of novel oily amino-pyrrolidinyl phosphonates (17a-h) which have not been reported before. In addition, they are thermally and air-stable compounds at room temperature, and are soluble in most organic solvents. Besides, this cyclization reaction is general for both aliphatic and aromatic primary amines as shown in Table 1. In addition, they are of potent biological activity precisely as antibiotics analogous to compounds 1-12 as shown in Table 2.
Unlike primary amines, when secondary amines were used, no heterocycles were detected and only 2-amino-cyclobutenylphosphonates 16 were obtained.
A suggested mechanism for this reaction can be attributed to initial addition of the amine on the carbon-carbon double bond to give a zwitterionic intermediate followed by proton transfer. Then, another hydroamination reaction took place on the double bond in the presence of excess amine to give the intermediate (20). After, nucleophilic attack of the nitrogen atom on C1 onto the carbon C-Cl by SN2 fashion, an aminopyrrolidinyl phosphonate (17) was produced (Scheme 3) [31] [32] [33] [34].
![]()
Table 1. Synthesis of pyrrolidinylphosphonates 17a-h. 
aAfter silica gel chromatography; b>98% as determined by GC/MS and 31P NMR.
![]()
Table 2. Predicted biological activity of compounds 1-17.
*Only activities with Pa > 0.5 are shown.
1H NMR (300 MHz, Chloroform d): δ 1.01 (d, 6H, JHH = 6.3 Hz), 1.11 (d, 6H, JHH = 6.3 Hz), 1.30 (t, 6H, JHH = 6.9 Hz), 1.65 - 2.30 (overlap, 3H), 2.65 - 70 (m, 1H), 2.78 - 2.88 (overlap, 2H), 3.02 - 3.15 (m, 1H), 3.42 - 3.48 (m, 1H), 4.03 - 4.12 (m, 4H); 31P NMR (121.4 MHZ, Chloroform d): δ31.38; 13C NMR (75.5 MHz, Chloroform d): 15.8 (d, 3JPC = 6.8), 15.8, 15.9, 32.0 (d, 1JPC = 126.7 Hz), 44.4, 50.8 (d, 2JPC = 3.2 Hz), 53.1, 60.4 (d, 2JPC = 8.6 Hz), 62.1, 63.2; MS(EI):m/z (%) 306 (10.5), 305 (15.1), 287 (20.0), 235 (22.3), 168 (35.0), 152 (18.8), 123 (30.0), 110 (41.3), 84 (100), 70 (56.7), 42 (19.9); Anal. Calcd. for C14H31N2O3P: C, 54.88; H, 10.20; N, 9.14; P, 10.11. Found: C, 55.09; H, 10.37; N, 8.95; P, 9.97.
1H NMR (300 MHz, Chloroform d): δ 1.09 (s, 9H), 1.11 (s, 9H), 1.31 (dt, 6H, JHH = 7.2 Hz, 3JHP = 2.1 Hz), 1.63 - 2.22 (overlap, 3H), 2.61 - 2.67 (overlap m, 2H), 3.21 - 3.45 (m, 1H), 4.06 - 4.13 (m, 4H); 31P NMR (121.4 MHZ, Chloroform d): δ 30.38; 13C NMR (75.5 MHz, Chloroform d): 16.4 (d, 3JPC = 6.3), 28.8, 30.0, 34.5 (d, 1JPC = 132.4 Hz), 39.7, 47.0, 50.6, 50.8 (d, 2JPC = 3.0 Hz), 60.4 (d, 2JPC = 8.6 Hz); MS(EI): m/z (%) 334 (0.70), 319 (2.5), 289 (7.8), 277 (17.8), 244 (27.8), 220 (70.5), 186 (51.1), 136 (100), 89 (55.1), 57 (67.8); Anal. Calcd. for C16H35N2O3P: C, 57.46; H, 10.55; N, 8.38; P, 9.26. Found: C, 58.18; H, 10.73; N, 8.21; P, 9.12.
1H NMR (300 MHz, Chloroform d): 1H NMR (300 MHz, Chloroform d): δ 1.26 (dt, 6H, JHH = 6.9 Hz, 3JHP = 1.51 Hz), 1.69 - 2.18 (overlap, 3H), 2.64 - 283 (m, 2H), 3.05 - 3.14 (m, 1H), 3.76 (s, 2H), 3.77 (s, 2H), 4.00 - 4.11 (m, 4H), 7.21 - 7.33 (overlap, 10); 31P NMR (121.4 MHZ, Chloroform d): δ 30.98; 13C NMR (75.5 MHz, Chloroform d): 16.1 (d, 3JPC = 7.4), 30.5, 40.8, 47.5 (d, 1JPC = 126.7 Hz), 45.5, 55.0, 60.2 (d, 2JPC = 4.0 Hz), 60.9 (d, 2JPC = 5.7 Hz), 127.7, 127.0, 128.2, 141.5; MS(EI): m/z (%) 402 (3.1), 401 (4.7), 325 (15.4), 311(17.7), 295 (17.9), 266 (30.4), 218 (44.9), 186 (22.3), 136 (60.1), 104 (33.6), 91 (100), 77 (55.8), 65 (18.8), 54 (26.8); Anal. Calcd. for C22H31N2O3P: C, 65.65; H, 7.76; N, 6.96; P, 7.70. Found: C, 65.47; H, 7.64; N, 7.10; P, 7.81.
1H NMR (300 MHz, Chloroform d): δ 0.89 (t, 3H, JHH = 5.7 Hz), 0.92 (t, 3H, JHH = 5.7 Hz), 1.31 (dt, 6H, JHH = 7.2 Hz, 3JHP = 2.1 Hz), 1.21 - 1.34 (overlap, 8H), 1.40 - 1.51 (m, 2H), 1.53 - 1.70 (m, 2H), 1.80 - 2.21 (overlap, 3H), 2.55 - 2.80 (overlap, 4H), 2.81 - 2.90 ((m, 1H), 3.00 - 3.05 (m, 1H), 3.06 - 3.15 (m, 1H), 4.06 - 4.15 (m, 4H); 31P NMR (121.4 MHZ, Chloroform d): δ31.47; 13C NMR (75.5 MHz, Chloroform d): 13.9, 14.1, 16.4 (d, 3JPC = 6.3), 22.4, 22.6, 26.4, 27.9, 29.2, 29.5, 29.8, 30.1 (d, 1JPC = 137.4 Hz), 32.3, 46.5, 47.1, 48.9 (d, 2JPC = 3.0 Hz), 55.4, 61.4 (d, 2JPC = 8.6 Hz); MS(EI): m/z (%) 362 (0.8), 319 (0.9), 279 (1.1), 264 (8.4), 256 (2.0), 222 (15.0), 194 (42.9), 142 (28.1), 128 (100), 98 (28.8), 82 (14.3), 56 (34.4), 43 (49.8), 29 (64.2); Anal. Calcd. for C18H39N2O3P: C, 59.64; H, 10.84; N, 7.73; P, 8.54. Found: C, 59.77; H, 10.97; N, 7.60; P, 8.41.
1H NMR (300 MHz, Chloroform d): δ 0.88 (t, 3H, JHH = 5.7 Hz), 0.96 (t, 3H, JHH = 5.7 Hz), 1.31 (dt, 6H, JHH = 7.2 Hz, 3JHP = 2.1 Hz), 1.35 - 1.48 (overlap, 10H), 1.60 - 2.17 (overlap, 7H), 2.90 - 3.10 (m, 1H), 4.03 - 4.15 (m, 4H); 31P NMR (121.4 MHZ, Chloroform d): δ 31.85; 13C NMR (75.5 MHz, Chloroform d): 13.6, 14.0, 16.2 (d, 3JPC = 6.5), 23.0, 23.8, 28.3, 29.8, 30.3, 31.5 (d, 1JPC = 140.2 Hz), 33.5, 48.1, 49.3 (d, 2JPC = 3.2 Hz), 50.0, 53.1, 62.0 (d, 2JPC = 8.4 Hz); MS(EI):m/z (%) 334 (0.9), 333 (1.0), 319 (16.8), 304 (40.2), 289 (17.6), 276 (66.1), 219 (32.2), 189 (100), 135 (47.8), 89 (41.0), 57 (44.1); Anal. Calcd. for C16H35N2O3P: C, 57.46; H, 10.55; N, 8.38; P, 9.26. Found: C, 57.63; H, 10.70; N, 8.19; P, 9.09.
1H NMR (300 MHz, Chloroform d): 1H NMR (300 MHz, Chloroform d): δ 1.22 (dt, 6H, JHH = 6.9 Hz, 3JHP = 2.1 Hz), 1.73 - 2.20 (overlap, 3H), 2.58 - 2.80 (m, 2H), 3.00 - 3.10 (m, 1H), 4.08 - 4.13 (m, 4H), 7.21 - 7.35 (overlap, 10); 31P NMR (121.4 MHZ, Chloroform d): δ 30.98; 13C NMR (75.5 MHz, Chloroform d): 16.2 (d, 3JPC = 6.8), 29.8, 39.9, 48.8 (d, 1JPC = 132.1 Hz), 47.0, 61.0 (d, 2JPC = 2.8 Hz), 62.5 (d, 2JPC = 5.7 Hz), 127.9, 128.8, 129.2, 141.0; MS(EI): m/z (%) 374 (0.7), 373 (1.2), 297 (30.0), 255 (20.1), 220 (100), 189 (35.9), 137 (28.7), 89 (18.7), 77 (67.9), 65 (30.0); Anal. Calcd. for C20H27N2O3P: C, 64.16; H, 7.27; N, 7.48; P, 8.27. Found: C, 63.98; H, 7.13; N, 7.60; P, 8.41.
The 1H, 13C, and 31P NMR spectra were recorded from solutions in CDCl3 on a Varian Mercury 300 spectrometer at 300, 75.5, and 121.4 MHz, respectively; the chemical shifts were measured relative to TMS (1H, 13C) and H3PO4. The mass spectra (EI) were recorded on an HP G1800A GCD GC/MS instrument using a 30-m methyl silicone column.
Synthesis of Diethyl (4-chlorobut-1-yn-1-yl)phosphonate (15)
Since the starting material 4-chlorobut-1-yne was not commercially available as usual, it was prepared in our lab [35].
Synthesis of aminopyrrolidinyl phosphonates (17a-h)
Typical procedure for the synthesis of diethyl (1-isopropyl-3-(isopropylamino)pyrrolidin-2-yl)phosphonate 17a.
To diethyl (4-chlorobut-1-yn-1-yl)phosphonate (0.22 g, 1 mmol) was added (0.23 g, 3.5 mmol) of isopropylamine in a 10 mL round bottom flask. After stirring at 25˚C for 12 h. the reaction mixture was washed with 0.1 N NaOH solution and the product was extracted with (2 × 20 mL CH2Cl2), dried over MgSO4, concentrated using a rotary evaporator and the oily product was separated on a silica gel column and was obtained in 80% isolated yield (10% methanol:90% dichloromethane), which was then analyzed by GC/MS, elemental analysis, and NMR spectroscopy.
1H NMR (300 MHz, Chloroform d): δ 1.01 (d, 6H, JHH = 6.3 Hz), 1.11 (d, 6H, JHH = 6.3 Hz), 1.30 (t, 6H, JHH = 6.9 Hz), 1.65 - 2.30 (overlap, 3H), 2.65 - 70 (m, 1H), 2.78 - 2.88 (overlap, 2H), 3.02 - 3.15 (m, 1H), 3.42 - 3.48 (m, 1H), 4.03 - 4.12 (m, 4H); 31P NMR (121.4 MHZ, Chloroform d): δ31.38; 13C NMR (75.5 MHz, Chloroform d): 15.8 (d, 3JPC = 6.8), 15.8, 15.9, 32.0 (d, 1JPC = 126.7 Hz), 44.4, 50.8 (d, 2JPC = 3.2 Hz), 53.1, 60.4 (d, 2JPC = 8.6 Hz), 62.1, 63.2; MS(EI):m/z (%) 306 (10.5), 305 (15.1), 287 (20.0), 235 (22.3), 168 (35.0), 152 (18.8), 123 (30.0), 110 (41.3), 84 (100), 70 (56.7), 42 (19.9); Anal. Calcd. for C14H31N2O3P: C, 54.88; H, 10.20; N, 9.14; P, 10.11. Found: C, 55.09; H, 10.37; N, 8.95; P, 9.97.
Synthesis of diethyl (1-(tert-butyl)-3-(tert-butylamino)pyrrolidin-2-yl)phosphonate 17b.
Identical to procedure 17a except adding t-butylamine and was obtained in 74% isolated yield.
1H NMR (300 MHz, Chloroform d): δ 1.09 (s, 9H), 1.11 (s, 9H), 1.31 (dt, 6H, JHH = 7.2 Hz, 3JHP = 2.1 Hz), 1.63 - 2.22 (overlap, 3H), 2.61 - 2.67 (overlap m, 2H), 3.21 - 3.45 (m, 1H), 4.06 - 4.13 (m, 4H); 31P NMR (121.4 MHZ, Chloroform d): δ 30.38; 13C NMR (75.5 MHz, Chloroform d): 16.4 (d, 3JPC = 6.3), 28.8, 30.0, 34.5 (d, 1JPC = 132.4 Hz), 39.7, 47.0, 50.6, 50.8 (d, 2JPC = 3.0 Hz), 60.4 (d, 2JPC = 8.6 Hz); MS(EI): m/z (%) 334 (0.70), 319 (2.5), 289 (7.8), 277 (17.8), 244 (27.8), 220 (70.5), 186 (51.1), 136 (100), 89 (55.1), 57 (67.8); Anal. Calcd. for C16H35N2O3P: C, 57.46; H, 10.55; N, 8.38; P, 9.26. Found: C, 58.18; H, 10.73; N, 8.21; P, 9.12.
Synthesis of diethyl(1-benzyl-3-(benzylamino)pyrrolidin-2-yl)phosphonate 17c.
Identical to procedure 17a except adding benzylamine and was obtained in 78% isolated yield.
1H NMR (300 MHz, Chloroform d): 1H NMR (300 MHz, Chloroform d): δ 1.26 (dt, 6H, JHH = 6.9 Hz, 3JHP = 1.51 Hz), 1.69 - 2.18 (overlap, 3H), 2.64 - 283 (m, 2H), 3.05 - 3.14 (m, 1H), 3.76 (s, 2H), 3.77 (s, 2H), 4.00 - 4.11 (m, 4H), 7.21 - 7.33 (overlap, 10); 31P NMR (121.4 MHZ, Chloroform d): δ 30.98; 13C NMR (75.5 MHz, Chloroform d): 16.1 (d, 3JPC = 7.4), 30.5, 40.8, 47.5 (d, 1JPC = 126.7 Hz), 45.5, 55.0, 60.2 (d, 2JPC = 4.0 Hz), 60.9 (d, 2JPC = 5.7 Hz), 127.7, 127.0, 128.2, 141.5; MS(EI): m/z (%) 402 (3.1), 401 (4.7), 325 (15.4), 311(17.7), 295 (17.9), 266 (30.4), 218 (44.9), 186 (22.3), 136 (60.1), 104 (33.6), 91 (100), 77 (55.8), 65 (18.8), 54 (26.8); Anal. Calcd. for C22H31N2O3P: C, 65.65; H, 7.76; N, 6.96; P, 7.70. Found: C, 65.47; H, 7.64; N, 7.10; P, 7.81.
Synthesis of diethyl (1-pentyl-3-(pentylamino)pyrrolidin-2-yl)phosphonate17d.
Identical to procedure 17a except adding amylamine and was obtained in 75% isolated yield.
1H NMR (300 MHz, Chloroform d): δ 0.89 (t, 3H, JHH = 5.7 Hz), 0.92 (t, 3H, JHH = 5.7 Hz), 1.31 (dt, 6H, JHH = 7.2 Hz, 3JHP = 2.1 Hz), 1.21 - 1.34 (overlap, 8H), 1.40 - 1.51 (m, 2H), 1.53 - 1.70 (m, 2H), 1.80 - 2.21 (overlap, 3H), 2.55 - 2.80 (overlap, 4H), 2.81 - 2.90 ((m, 1H), 3.00 - 3.05 (m, 1H), 3.06 - 3.15 (m, 1H), 4.06 - 4.15 (m, 4H); 31P NMR (121.4 MHZ, Chloroform d): δ31.47; 13C NMR (75.5 MHz, Chloroform d): 13.9, 14.1, 16.4 (d, 3JPC = 6.3), 22.4, 22.6, 26.4, 27.9, 29.2, 29.5, 29.8, 30.1 (d, 1JPC = 137.4 Hz), 32.3, 46.5, 47.1, 48.9 (d, 2JPC = 3.0 Hz), 55.4, 61.4 (d, 2JPC = 8.6 Hz); MS(EI): m/z (%) 362 (0.8), 319 (0.9), 279 (1.1), 264 (8.4), 256 (2.0), 222 (15.0), 194 (42.9), 142 (28.1), 128 (100), 98 (28.8), 82 (14.3), 56 (34.4), 43 (49.8), 29 (64.2); Anal. Calcd. for C18H39N2O3P: C, 59.64; H, 10.84; N, 7.73; P, 8.54. Found: C, 59.77; H, 10.97; N, 7.60; P, 8.41.
Synthesis of diethyl (1-butyl-3-(butylamino)pyrrolidin-2-yl)phosphonate 17e.
Identical to procedure 17a except adding butylamine and was obtained in 77% isolated yield.
1H NMR (300 MHz, Chloroform d): δ 0.88 (t, 3H, JHH = 5.7 Hz), 0.96 (t, 3H, JHH = 5.7 Hz), 1.31 (dt, 6H, JHH = 7.2 Hz, 3JHP = 2.1 Hz), 1.35 - 1.48 (overlap, 10H), 1.60 - 2.17 (overlap, 7H), 2.90 - 3.10 (m, 1H), 4.03 - 4.15 (m, 4H); 31P NMR (121.4 MHZ, Chloroform d): δ 31.85; 13C NMR (75.5 MHz, Chloroform d): 13.6, 14.0, 16.2 (d, 3JPC = 6.5), 23.0, 23.8, 28.3, 29.8, 30.3, 31.5 (d, 1JPC = 140.2 Hz), 33.5, 48.1, 49.3 (d, 2JPC = 3.2 Hz), 50.0, 53.1, 62.0 (d, 2JPC = 8.4 Hz); MS(EI):m/z (%) 334 (0.9), 333 (1.0), 319 (16.8), 304 (40.2), 289 (17.6), 276 (66.1), 219 (32.2), 189 (100), 135 (47.8), 89 (41.0), 57 (44.1); Anal. Calcd. for C16H35N2O3P: C, 57.46; H, 10.55; N, 8.38; P, 9.26. Found: C, 57.63; H, 10.70; N, 8.19; P, 9.09.
Synthesis of diethyl (1-phenyl-3-(phenylamino)pyrrolidin-2-yl)phosphonate 17f.
Identical to procedure 17a except adding phenylamine and was obtained in 76% isolated yield.
1H NMR (300 MHz, Chloroform d): 1H NMR (300 MHz, Chloroform d): δ 1.22 (dt, 6H, JHH = 6.9 Hz, 3JHP = 2.1 Hz), 1.73 - 2.20 (overlap, 3H), 2.58 - 2.80 (m, 2H), 3.00 - 3.10 (m, 1H), 4.08 - 4.13 (m, 4H), 7.21 - 7.35 (overlap, 10); 31P NMR (121.4 MHZ, Chloroform d): δ 30.98; 13C NMR (75.5 MHz, Chloroform d): 16.2 (d, 3JPC = 6.8), 29.8, 39.9, 48.8 (d, 1JPC = 132.1 Hz), 47.0, 61.0 (d, 2JPC = 2.8 Hz), 62.5 (d, 2JPC = 5.7 Hz), 127.9, 128.8, 129.2, 141.0; MS(EI): m/z (%) 374 (0.7), 373 (1.2), 297 (30.0), 255 (20.1), 220 (100), 189 (35.9), 137 (28.7), 89 (18.7), 77 (67.9), 65 (30.0); Anal. Calcd. for C20H27N2O3P: C, 64.16; H, 7.27; N, 7.48; P, 8.27. Found: C, 63.98; H, 7.13; N, 7.60; P, 8.41.
Synthesis of diethyl (1-heptyl-3-(heptylamino)pyrrolidin-2-yl)phosphonate 17g.
Identical to procedure 17a except adding heptylamine and was obtained in 75% isolated yield.
1H NMR (300 MHz, Chloroform d): δ 0.88 (t, 3H, JHH = 5.7 Hz), 0.96 (t, 3H, JHH = 5.7 Hz), 1.28 (dt, 6H, JHH = 7.2 Hz, 3JHP = 2.1 Hz), 1.30 - 1.60 (overlap, 16H), 1.74 - 2.18 (overlap, 13H), 2.93 - 3.20 (m, 1H), 4.04 - 4.10 (m, 4H); 31P NMR (121.4 MHZ, Chloroform d): δ 32.25; 13C NMR (75.5 MHz, Chloroform d): 13.6, 14.0, 16.0 (d, 3JPC = 6.1), 22.2, 22.7, 26.9, 27.0, 27.9, 28.2, 28.8, 29.0, 29.4, 29.8, 31.0 (d, 1JPC = 135.8 Hz), 33.3, 45.2, 46.9 (d, 2JPC = 3.2 Hz), 49.0, 53.0, 60.8 (d, 2JPC = 8.6 Hz); MS(EI): m/z (%) 418 (0.7), 417 (0.9), 412 (11.2), 396 (14.8), 345 (20.7), 289 (49.6), 235 (33.8), 189 (100), 135 (60.3), 87 (57.7), 43 (45.8); Anal. Calcd. for C22H47N2O3P: C, 63.12; H, 11.32; N, 6.69; P, 7.40. Found: C, 62.91; H, 11.17; N, 6.81; P, 7.56.
Synthesis of diethyl (1-phenethyl-3-(phenethylamino)pyrrolidin-2-yl)phosphonate17h.
Identical to procedure 17a except adding phenylethylamine and was obtained in 79% isolated yield.
1H NMR (300 MHz, Chloroform d): 1H NMR (300 MHz, Chloroform d): δ 1.24 (dt, 6H, JHH = 6.9 Hz, 3JHP = 1.67 Hz), 1.58 - 2.18 (overlap, 3H), 2.14 - 2.85 (overlap, 7H), 4.04 - 4.15 (m, 4H), 7.17 - 7.31 (overlap, 10); 31P NMR (121.4 MHZ, Chloroform d): δ 31.78; 13C NMR (75.5 MHz, Chloroform d): 16.2 (d, 3JPC = 7.2), 31.3, 32.8, 41.5, 49.5 (d, 1JPC = 132.9 Hz), 48.2, 55.6, 57.9, 61.0 (d, 2JPC = 4.2 Hz), 60.9 (d, 2JPC = 5.7 Hz), 127.7, 127.0, 128.2, 141.5; MS(EI): m/z (%) 430 (0.7), 429 (0.8), 353 (18.9), 325 (32.1), 276 (100), 234 (22.8), 188 (45.9), 135 (57.7), 105 (77.8), 77 (46.8), 69 (66.7); Anal. Calcd. for C24H35N2O3P: C, 66.96; H, 8.19; N, 6.51; P, 7.19. Found: C, 67.08; H, 8.31; N, 6.37; P, 7.06.
Acknowledgements
This research was supported by Al Quds University funding (AAQ, HD, AJ). The work (GTA) was performed in the framework of the Program for Basic Research of State Academies of Sciences for 2013-2020, also this work (DVM) was supported by the Russian Science Foundation (Grant No. 18-73-10030).