1. Introduction
Platinum features an outstanding catalytic activity for various reactions, in particular the oxygen reduction reaction (ORR), and the oxidation of the ethanol, and the methanol. These three reactions are key steps in the working cycle of proton exchange membrane fuel cells (PEFMCs) and direct methanol fuel cells (DMFCs), which represent promising alternatives to fossil fuels as a power source for vehicles. The performances of the catalysts based on Pt can be improved by the incorporation of nanostructures with higher active surface areas and, hence, higher efficiencies regarding the targeted chemical reactions. Consequently, different types of nanoparticles were synthesized possessing different sizes and shapes: Nanocubes [1] , nanospheres [2] and bimetallic particles, like Pt-Cu [3] , Pt-Pd [4] , Pt-Ni [5] , which enable the enhancement of the catalytic activity and also the reduction of the required amount of expensive platinum. The bimetallic particles are, e.g., alloys [6] , core-shell particles [7] , or nanocages [8] . In this work, we study Pt-Pd core-shell nanoparticles that are synthesized by galvanic replacement applying varying synthesis times. Different studies on Pt- Pd nanoparticles and their catalytic activity regarding the ORR were already realized, but we intended to monitor the evolution of the surface of the particles during the different steps of the reaction, namely the adsorption of oxygen and formation of oxides, through the monitoring of the Pt-Pd and Pt-O bond. Here, the in-situ XAFS technique represents a powerful method to study the surface of the particles during the ORR [1] [9] [10] . However, for the in-situ Extended X-ray absorption fine structure (EXAFS) and X-ray absorption near edge structure (XANES) experiments, the standard fitting approach for the analysis of the spectra is not always suited to separate the different species that are present in the system. Thus, we analyzed our results using the Iterative Transformation Factor Analysis (ITFA) [11] , which enabled the comparison of the different particles via their spectra and catalytic properties.
2. Experimental Section
2.1. Materials and Methods
Sodium tetrachloropalladate (Na2PdCl4, 98%), potassium tetrachloroplatinate (K2PtCl4, 98%), ascorbic acid (AA, 98%), potassium bromide, (KBr, 99%), Nafion®, perfluorinated resin solution (5 wt.%) and nitric acid (HNO3, 70%, purified by distillation) were purchased from Sigma Aldrich. Isopropyl alcohol, (IPA, 99.5%) was purchased from Merck, ethylene glycol from VWR and poly (vinylpyrrolidone) (PVP, 98%) from Alfa Aesar. The black carbon XC-72R was purchased from Vulcan and the microfiltration membrane (0.45 μm filter) from Diapore, the plain carbon cloth (model 1071) from FuelCellsEtc and the graphite conductive adhesive, alcohol-based (42,465) from Alpha Aesar.
2.2. Synthesis of PtPd Nanoparticles
Depending on the particle type, size, shape, and composition, different methods were used to synthesize PdPt nanoparticles (NPs) in aqueous or organic solvents [12] [13] [14] . The synthesis of bimetallic core-shell nanoparticles [15] and PdPt alloy nanocages [8] was based on the work by Zhang et al. [15] . Hexachloroplatinic acid (H2PtCl6) was replaced by potassium tetrachloroplatinate (K2PtCl4), which possesses a lower oxidation state for the platinum (Pt(II)).
The galvanic replacement method to synthesize the particles was performed in two steps: First, the synthesis of Pd cubic nanoparticles, followed by the Pt-Pd core-shell nanoparticles. The Pd NPs were prepared in a solution of 0.6 g of ascorbic acid, 3 g of potassium bromide, 1.85 g of potassium chloride, and 1.05 g of poly (vinylpyrrolidone), in 80 mL of distilled water. After sonication (2 min), the solution was heated to 80˚C. After 10 min, 30 mL of Na2PdCl4 (6.5 × 10−2 mol∙L−1) was added with a syringe (1 mL∙min−1). Afterwards, the solution was kept for 3.5 h at 80˚C under magnetic stirring with a rotational speed of 1200 rpm. The platinum was added in a second step to form the PdPt nanoparticles: 10 mL of the solution containing Pd nanoparticles were mixed with 70 mL of an aqueous solution containing 3 g of KBr and 0.333 g of PVP. After heating to 90˚C, 30 mL of K2PtCl4 (3 × 10−3 mol∙L−1) were added (1 mL∙min−1) under magnetic stirring (1200 rpm). Three synthesis durations were used to study the evolution of the particles and their performances: 0.6 h, 2 h and 8 h respectively. Subsequently, the particles were collected by centrifugation, washed several times with water as well as ethanol and dried in an oven at 70˚C.
2.3. Preparation of the Particles for the In-Situ XAFS and Electrochemical Measurements
To study the particles via in-situ XAFS and electrochemical means, a mixture with carbon applying a particles-carbon weight ratio of 2:3 was used. The same method was used to coat the particles on carbon for all samples: A solution with 40 mg of particles in 25 mL of ethylene glycol was sonicated for 15 min and a second solution containing 60 mg of carbon Vulcan XC-72R powder diluted in 25 mL of ethylene glycol adjusted at pH 2 (with nitric acid solution, 2 mol∙L−1) was sonicated for 30 min. Then, both solutions were mixed, sonicated for 1 h, and kept one night under magnetic stirring. Afterwards, the mixture was heated to 40˚C and filtrated. Microfiltration was used to collect the particles coated on carbon, and they were dried for 5 h at 70˚C in an oven. After coating on carbon, an ink was created by addition of 9 mg of particles coated on carbon in a solution containing 0.25 mL of water and 2.25 mL of isopropyl alcohol.
2.4. Electrochemical Measurement of the Catalytic Activity
All measurements were carried out in an electrochemical cell containing HClO4 (1 × 10−1 mol∙L−1) electrolyte with a platinum wire as counter electrode, a Hg/HgSO4 (0.654 V vs. standard hydrogen electrode (SHE) in saturated K2SO4) reference electrode and a rotating disk electrode (Princeton Applied Research, RDE 0008) with a geometric area of 0.196 cm−2 (diameter of 5 mm) as the working electrode. Before the measurements, the glassy carbon disk was polished and rinsed with deionized water, then cleaned ultrasonically in ultra-pure water for 5 min, and rinsed with isopropanol and acetone. After 15 min of sonication, 16 μL of ink containing the particles coated on carbon was deposited on the glassy carbon disk, covered by 7 μL of a Nafion® solution (volumetric ratio of Nafion® to methanol was 1:100), and all was dried under ambient conditions. Prior to the measurement of the ORR, the electrode conditioning was conducted by cyclic voltammetry from 0.050 to 1.200 V (all the potentials are versus the SHE) with a scan rate of 0.100 V∙s−1 until reproducible voltammograms were obtained. The electrochemical performance for the oxygen reduction reaction (ORR) was determined using linear scan voltammetry (LSV) applied to the rotating disk electrode in the potential range from 0.300 to 1.200 V with a scan rate of 5 × 10−3 V∙s−1 and a rotational speed of 1600 rpm in an O2-saturated electrolyte.
2.5. Characterization via TEM-EDS
The nanoparticles were studied using a high-resolution transmission electron microscope (HR-TEM) JEOL JEM-3010 operating at 300 keV and equipped with an X-Ray analyzer Oxford to perform energy dispersive spectroscopy (EDS) analysis. Just before the measurement, the homogenization of the aqueous solution containing nanoparticles was done by sonication. Subsequently, one drop of solution was deposited on a TEM grid (copper covered by a carbon film) and dried under vacuum.
2.6. XAFS Measurements
All XAFS experiments were performed at the Rossendorf Beamline (BM20A), European Synchrotron Radiation Facility (ESRF), Grenoble, France. A water- cooled Si (111) double-crystal monochromator (DCM) was used to monochromatize the synchrotron beam. Before the DCM, collimation was done with a 1.4 m long, Pt-coated, meridionally bent silicon mirror. A second, 1.3 m long, Pt- coated, toroidal silicon mirror focused the beam vertically and horizontally after the DCM. The rejection of higher harmonics of the complete setup is better than 104. The measurements, in fluorescence mode, were done at room temperature using ionization chambers and a 13-elements high-purity germanium detector (Canberra) with a digital spectrometer (XIA X-Map).
For the XAFS measurements, pellets that were prepared from 1 g of particles coated on Vulcan XC72-R carbon powder (40wt.-% particles) and boron nitride for a higher stability were used. The particles were characterized using the Pt-LIII, and Pd-K edges, providing information about the Pt-Pt (Pt-LIII edge), the Pd-Pd (Pd-K edge), and the Pt-Pd bond (Pt-LIII and Pd-K edge).
In-situ measurements were performed using a three electrodes system with a home-made cell filled with 8 mL of an HClO4 (1 × 10−1 mol∙L−1) aqueous solution. The counter electrode was a plain carbon cloth (0.5 cm × 2.5 cm) covered by conductive graphite, and an Ag/AgCl electrode was used as reference.
The working electrode was a plain carbon cloth covered by conductive graphite and was used to study the particles: 100 μL of the ink were deposited on the working electrode and then protected through the deposition of 16 μL of Nafion solution (ratio 1:100 Nafion: methanol). The area of the working electrode in solution was 0.5 cm2. After 5 min of monitoring the open circuit potential and 80 cycles from 0.05 to 1.10 V at 0.1 V∙s−1, the measurements were done at different potentials: 0.65, 0.85 V (oxidation), 1.20 V (vertex point), 1.00, 0.85, 0.65 and 0.35 V (re-reduction) (Figure 1). For all measurements, the energy calibration, based on the average of several scans, and the dead-time correction of the fluorescence signals were done with the SixPack [16] software. Subsequent data reduction steps were performed with WinXAS [17] . For the normalization of the spectra, a first-order polynomial function was fitted to the pre-edge region, and a second-order polynomial function was fitted to the post-edge region.
Subsequently, the conversion from energies to photoelectrons kinetic energy units (k-space) was performed by arbitrarily assigning the first inflexion point of the absorption edge to zero energy. The auto-spline algorithm of WinXAS permitted the extraction of the EXAFS oscillations from the post-edge region by a spline fit. After a k3-weighting, the obtained EXAFS functions were Fourier-transformed to the R space using a Bessel window function across the k range from 2 to 12 Å−1. The theoretical phase shift and amplitude functions for the shell fits were calculated using the FEFF 8.2 source code [18] based on Pt, Pt-Pd and Pd metal clusters with face-centered cubic (fcc) structure. The shell fit was performed with WinXAS to derive coordination numbers (CN), interatomic distances (R), Debye-Waller factors (σ2), and phase shift corrections (ΔE0).
2.7. Analysis of Spectra via Principal Component Analysis, Varimax, and Iterative Target Transformation Factor Analysis
The evolution of the in-situ XAFS spectra was studied using a program developed by A. Rossberg, A. C. Scheinost et al. [19] [20] , permitting the analysis of the XAFS spectra via the Iterative Transformation Factor Analysis (ITFA) with a calculus of Varimax and Iterative Target Test (ITT). The first step of the ITFA is the Principal Component Analysis (PCA) [21] . The goal of the PCA is to identify the number of independent components that are required to reproduce the experimental spectrum. The work-flow uses the theoretical root mean square functions developed by Malinowski [22] : The real error (RE), the embedded error (IE), and the semi-empirical indicator (INA). All these functions depend on the
![]()
Figure 1. Standard ORR cycle and points used for the in-situ XAFS measurements.
number of components. Firstly, the eigenvectors and the factor logins have to be set, representing the n-dimensional factor space with an orthogonal basis formed by the eigenvectors. For one sample, with seven spectra, the PCA is carried out with a factor loading of seven, the maximum number of components necessary to reconstruct the spectra. With the reduction of the dimensional factor, differences between the spectra, corresponding to the different components, become apparent. Finally, differences with a dimensional factor of two were found: Two main spectra (corresponding to two different components) permitted the reconstruction of the experimental spectrum of one sample. The other spectra are attributed to the experimental error and the noise. After the PCA and the reconstruction of the spectra, the evolution of the relative concentrations of the main components visible for the samples is followed using the Varimax method and the Iterative Target Test (ITT). The Varimax method, a statistical rotation method developed by H. F. Kaiser [23] , aims at preferably high or low loadings for the factors of the linear combinations that resemble the different experimental spectra by rotation of the underlying n-dimensional space. Thus, an assignment of the single components of the spectra to the different spectra and, hence, the interpretation of the data becomes easier. Namely, applying the Varimax method allows the identification of correlations of the spectra with the components. Using the spectra with the highest or lowest factor loadings, the components were identified and the ITT enabled the calculation of their relative concentrations.
3. Results and Discussion
3.1. TEM Images and EDS Measurements
The evolution of the shape of the particles, in particular their concavity, was monitored using TEM imaging (Figure 2). After 0.6 h, particles with a size of ca. 18 nm and a cubic shape were obtained. The formation of very thin layers that cover the cores of the cubes is apparent (darker edges in the TEM image).After 2 h, the particles feature a concave shape and contain more platinum, as evidently from the lower contrast between the core and the external thicker layer. For the longest synthesis time, 8 h, the particles contain more platinum, but the shapes
![]()
Figure 2. TEM images of 1) PtPd 0.6 h (left), PtPd 2 h (middle), PtPd 8 h (right).
of the particles are only negligibly different from those obtained after 2 h. However, additional larger, hexagonal structures with a small cubic, dark core (~10 nm) covered by a lighter layer to form hexagons were observed. With EDS measurements on different particles, from the dark core to the shell, an evolution of the chemical composition for the different particles is visible, with a higher concentration of Pt in the shell (lighter layer). These observations suggest the external layer is mainly constituted of platinum, while the core consists of palladium with a small amount of platinum. The particles mainly consist of palladium and show an increasing amount of platinum from 9% to 26% with increasing reaction time (Table 1). One high resolution EDS mapping can help to confirm this analysis.
3.2. EXAFS Measurements at the Pt-LIII and Pd-K Edges
After the Fourier transformation of the EXAFS spectra that were collected at the Pt-LIII edge, a well-defined double peak between 2 and 3 Å becomes apparent for all the particles (Figure 3). Modeling with FEFF using Pt and Pd clusters with
![]()
Table 1. Evolution of the amount of Pt and Pd in the particles measured by EDS.
![]()
Figure 3. Fourier-transformed EXAFS spectra (uncorrected shift) of nanoparticles: 0.6 h (blue), 2h (red), 8 h (green) at the Pt-LIII and Pd-K edges.
![]()
Table 2. Coordination numbers (CN), interatomic distances (R), Debye-Waller factors (σ2), and energy shifts (ΔE0) achieved from the Pt-LIII edge analysis.
![]()
Table 3. Coordination numbers (CN), interatomic distances R, Debye-Waller factors (σ2), and energy shifts (ΔE0) achieved from the Pd-K edge analysis.
different elemental ratios allows the attribution of this double peak to the presence of neighboring Pt and Pd in similar proportions in the first shell. Compared to the nanoparticles spectra, only one peak is visible for the Pt foil, which is in agreement with particles with only one type of neighbors, namely the Pt atoms. In the Fourier-transformed Pd-K-edge spectra, the single main peak (R ~2.74 Å) suggests the predominance of only one type of neighboring atoms, most likely palladium.
The analysis of the obtained data with WinXAS software using models based on FEFF confirmed the first conclusions drawn from the experimental EXAFS spectra (Table 2).
The hypothesis of Pt atoms surrounded by platinum and palladium neighbors, based on the Pt-LIII edge spectra, is verified by the resulting equal distances between the Pt atoms and their neighbors of 2.72 to 2.74 Å.
These values are in line with the crystallographic data [24] . The Pt/Pd ratio is close to 1 and the sum of Coordination Numbers (CN) of Pt-Pt and Pt-Pd is close to and the sum of Coordination Numbers (CN) of Pt-Pt and Pt-Pd is close to 12 (considering an error of 25% for the CN).
With these results and the small Debye-Waller factors, indicating a small static and vibrational disorder, a shell possessing an fcc structure where platinum is surrounded by platinum and palladium atoms is most likely.
The analysis of the Pd-K edge spectra also yielded Pd-Pd distances for the nanoparticles and for the reference of 2.74 Å, which is in agreement with theoretical values for the fcc structure [24] . The Pd-Pt interatomic distance is significantly smaller than the Pd-Pd bond (Table 3).
Compared to the Pt atoms, the coordination numbers, between 10.2 and 11.2 for the Pd-Pd bond, suggest the predominance of Pd atoms neighbored by other Pd atoms and only a small number of Pt atoms (CN between 0.6 and 1.7). The different values determined by the fitting of the EXAFS curves for both Pd-K and Pt-LIII edges suggest that the nanoparticles possess a well-structured metallic core of palladium where most of the Pd atoms have Pd neighbors, surrounded by a thin shell of Pt atoms where most of Pt atoms still have Pd neighbors.
3.3. In-Situ XAFS Studies
The modifications of the structure and composition of the PtPd nanoparticles with changing potential were studied. For every sample, spectra were measured at the characteristic potentials of the ORR cycle (cf. Figure 1) resulting in seven spectra per sample. After the in-situ measurements and the dead-time corrections using the SixPack software, the spectra were normalized and the EXAFS functions were k3-weighted. Afterwards, the spectra were analyzed using Iterative Transformation Factor Analysis (ITFA).
By comparison with EXAFS standard spectra and modeling with FEFF, the two components are assigned to the corresponding structures and chemical species, namely to a component with a Pt-O bond and to a component based on a Pt-Pt or Pt-Pd bond, which possess rather similar values. New in-situ EXAFS measurement at the Pd-K edge could provide more information about the Pd-Pd and Pd-Pt bond.
The experimental spectra were reproduced applying the ITFA (Figure 4) with a combination of the two components (except for PtPd at 8 h) using a FEFF model, with a higher weight on the first component, suggesting a combination of Pt-O and Pt-Pd bonds within the samples. Regarding the measurements of Pt-Pd for the particles synthesized in 8 h, the experimental spectra can be reproduced with only one component, suggesting the presence of only one component and an insignificant amount of the second one, which is most likely the Pt-O assigned one. The coordination numbers (CN), interatomic distances (R), Debye-Waller factors (σ2), and energy shifts (ΔE0) are presented in Table 4, the analysis of the spectra is shown in Figure 4. The FEFF model is suitable for the analysis of the
![]()
Figure 4. EXAFS spectra forthe Pt-Pd nanoparticles at different synthesis times at 0.35 V: 0.6 h (blue), 2 h (red), 8 h (green).
![]()
Table 4. Coordination numbers (CN), interatomic distances R, Debye-Waller factors (σ2), and energy shifts (ΔE0) from the Pt-LIII edge spectra at 0.35 V determined by curve fitting.
experimental data with only small residuals. The small Debye-Waller factors indicate insignificant vibrational and static disorder, pointing particles with a well-defined structure. The determined values (Table 4) are similar to the values achieved from the ex-situ XAFS spectra (Table 2 and Table 3). Notably, for the ex-situ and, the in-situ EXAFS spectra at 0.35 V, the peak that corresponds to the Pt-O bond (R~ 1.8 Å) is not visible and the spectra can be fitted assuming only Pt, Pt-Pd and Pd metal clusters with face-centered cubic (fcc) structure using WinXAS software. Thus, the absence of oxide at an applied potential of 0.35 V is confirmed.
3.4. Evolution of the Structure with the Applied Potential
After the PCA, the reconstruction of the spectra (XANES and EXAFS) using two principal components (Pt metal and Pt oxide) allows the monitoring of the evolution of the samples with changing potential (Figure 5). The FEFF-based model of a Pt oxide cluster yields a peak at ~2 Å, corresponding to a theoretical length of the Pt-O bond of 2 Å.
For all samples (excluding the PtPd 8-h NPs), the Fourier-transformed EXAFS spectra feature a peak at R ~1.8 Å, which appears and disappears with the different potentials, while the inverse behavior occurs for the double peak between 2 and 3 Å, which is attributed to the Pt-Pd and Pt-Pt bond (only Pt-Pt for the commercial platinum). From 0.85 to 0.35 V, the single peak vanishes, and it reappears when the potential reaches 0.85 V again. Comparing the results with the Pt oxide model achieved from FEFF and the EXAFS database, this peak was assigned to the Pt-O bond. This suggests the formation of a thin layer of Pt oxide with oxygen atoms adsorbed on the surface of the particles when the potential is higher than 0.85 V. At 1.0 V and at 1.2 V (Figure 5), the peak corresponding to the Pt-O bond appears very distinctively in the spectra, except for the 8 h NP. These results are confirmed by the analysis of the experimental EXAFS curves using an FEFF model with Pt-Pt, Pt-Pd, and Pt oxide clusters (Table 5). For the commercial Pt, two Pt-Pt bonds were used for the curve fitting: One Pt-Pt bond (path 1) for pure Pt nanoparticles and a second one (path 2) corresponding to the Pt nanoparticles with Pt oxide. The results of the curve fitting (Table 5), including the Pt oxide in the model, are validated by small residual factors. Like for the EXAFS spectra measured at 0.35 V, the particles are well structured,
![]()
Figure 5. Experimental (black) and reconstruction (red) of XANES (left), EXAFS (insert) and Fourier transformed EXAFS spectra for the different potentials: (1) 0.6 h, (2) 2 h,(3) 8 h, (4) Pt commercial.
![]()
Table 5. Structural parameters of the particles at 1 V, Pt-LIII.
with small Debye-Waller factors. For the Pt-Pt shell, these factors are higher at 1.00 V than at 0.35 V for all the particles (excepted for the 8 h NPs), which could be due to the presence of Pt oxide, causing a disorder in the shell structure of the particles.
The lengths of the Pt-O bond, close, (~1.99 Å), are close to the theoretical value (2.07 Å) but the CN are smaller (CN = 6 in theory for pure Pt oxide). This is most likely due to the Pt oxide present at 1.00 V, unstructured compared to the Pt and Pd atoms well organized in a fcc crystalline system. Considering the experimental errors, the lengths of the Pt-Pt and PtPd bonds are close to the theoretical values (2.807 Å for the Pt-Pt bond and for the Pt-Pd bond) and to the bond length at 0.35 V. For the PtPd nanoparticles after 0.6 h and 2 h, the double peak shows a slight asymmetry with a higher peak height at 2.2 Å, which occurs for small potentials and disappears progressively with increasing potential. This was also observed by other researchers working on PtPd nanoparticles [25] . For the PtPd nanoparticles at 8 h, no significant changes are present and the spectra can be reproduced with only one component. Thus, due to the lack of any change with changing potential, the formation of an oxide layer is highly unlikely for these particles.
3.5. Studying the Relative Concentrations with the ITFA
For the systems based on two components, these were assigned to the oxide and the metal and the changes of their relative concentrations with changing potential were examined. The concentrations were normalized using the achieved data of the commercial Pt sample, presuming that only one metallic component is present and that the surface is constituted of pure platinum metal when the applied potential is 0.35 V and of platinum oxide. when the potential is 1.00 V, as it is stated in the related literature [26] [27] [28] .
Like that, the evolution of the relative concentrations (Figure 6) was monitored: When the potential is decreasing, the concentration of metal is increasing, while, in contrast, the concentration of the oxide is increasing with increasing potential, which was already observed in former studies on Pt and PtPd nanoparticles [25] . Obviously, the PtPd NP after 8 hours, whose spectra are based only on one component, cannot be analyzed using this model.
3.6. The ORR: Different Chemical Processes
For the oxygen reduction reaction, two main mechanisms are suggested: An as-
![]()
Figure 6. Evolution of the relative concentrations with the potential.
sociative and a dissociative one [26] [28] [29] .
Dissociative ORR mechanism
 (1)
(1)
 (2)
(2)
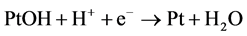 (3)
(3)
Associative ORR mechanism
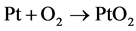 (4)
(4)
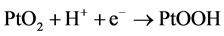 (5)
(5)
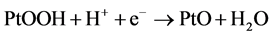 (6)
(6)
 (7)
(7)
![]() (8)
(8)
The first step of the associative mechanism is the adsorption of oxygen at the active sites of the electrode conserving the O-O bond, in contrast to the dissociative mechanisms, where the bond between the oxygen atoms is broken before the adsorption on the electrode. The last steps of both mechanisms (Equations ((2), (3), (7), and (8))) are equal and, at higher potentials, both mechanisms can occur. As already stated, above 0.85 V, the XAFS in-situ measurements suggest the presence of Pt-O bonds. Up to 1.00 V, the amount of oxide is increasing accompanied by a high degree of surface oxidation [30] , the relative concentration of the oxide determined using the Varimax method for our experimental data analysis is close to 1 while it is 0 for the metal.
3.7. Catalytic Activity of the Particles
The mass activity (MA), relative to the mass of catalyst loaded for the electrochemical oxygen reduction, was determined using linear sweep voltammetry with a rotating disk electrode (RDE). The rotation of the electrode ensures fast and effective convective transport of the electroactive species. The MA is related to the kinetic current density jk at 0.85 V, where the electrochemical processes are controlled by the electron transfer. According to the Koutecký-Levich equation, jk is defined by:
![]() (9)
(9)
where j is the total current density (measured at 0.85 V) and jd the diffusion-controlled current density (measured at 0.40 V). The mass activity (MA) is calculated from the kinetic current density relative to the mass of the nanoparticles mNP deposited at the surface of the RDE:
![]() (10)
(10)
The resulting mass activities of the different samples are summarized in Table 6.
The achieved mass activities are similar to reported values for Pt and Pt-Pd nanoparticles used as catalyst for the ORR [31] [32] . According to the experi-
![]()
Table 6. Catalytic activity for the different samples.
mental results, the Pt-Pd nanoparticles synthesized in 2 h represent the best system for the ORR catalysis, with a mass activity superior to the commercial Pt, and the performances of the particles synthesized in 0.6 h are rather close. Compared to the other samples, the 8 h NPs show a lower catalytic activity. With a smaller amount of platinum than the pure powder, the PtPd NPs at 0.6 h and the PtPd NPs at 2 h feature a good performance as catalysts for the ORR. The cubic shape of the particles and the Pt-Pd alloy on the surface of the particles may be the reason for their catalytic performance, with a higher electrochemical surface area and a beneficial composition of the alloy (Pt/Pd ratio) for the ORR.
Similar results have been found by other researchers suggesting an influence of the composition of the alloy [14] [33] .
4. Conclusion
The behavior and performance regarding the oxygen reduction reaction (ORR) was studied for different PtPd nanoparticles by means of electrochemical and in-situ XAFS measurements. For the latter, different electrochemical potentials were applied to reveal differences between the XAFS spectra of the different ORR steps. Iterative Transformation Factor Analysis (ITFA) was used to analyze the achieved experimental data. As the first step, the Principal Component Analysis (PCA) of the spectra, showed two main components, Pt or Pt-Pd in a metallic and in an oxide form, which allowed the reproduction of the experimental spectra. Continuing the ITFA using the Varimax method and the Iterative Target Test (ITT), the evolution of the relative concentrations of the two different forms of the particles was monitored. The oxide appears at potentials from 0.65 V and, is the main form up to 1.00 V. For the metallic species, the relative concentration is increasing with decreasing potentials down to 0.35 V, where it is the major component. The development of the structural parameters (CN, σ2, R) was calculated by analysis of the measured curves using WinXAS with Pt-Pd and α-PtO models that had been achieved through the FEFF code. The resulting values are in agreement with the results of the ITFA. Future in-situ XAFS measurements at the Pd-K edge would be helpful to gain information about the formation of Pd oxide during the ORR. Linear scanning voltammetry was used to evaluate the electrochemical performance of the different particles. However, characterization of the particles with other methods (X-ray photoelectrons spectroscopy, impedance spectrometry) is required to receive more details and more meaningful correlations between the nanoparticles’ structures and their catalytic activities.
Acknowledgements
We thank the research group working at the Rossendorf Beamline BM20A in the ESRF (Grenoble, France), Dr. Stephanie Höppener for EDS mapping, Dr. Igor Perevyazko for his help and access to a high-speed centrifugation, and the workshop of the university for the participation in the fabrication of the experimental cell. This work was carried out with the financial support of the Carl Zeiss Foundation (Germany).