Molecular Docking of the Inhibitory Activities of Triterpenoids of Lonchocarpus cyanescens against Ulcer ()
1. Introduction
Ulcers are holes that form in the lining of the upper part of the gastrointestinal tract (GIT). The different types of ulcer are peptic, gastric, and duodenal ulcers. A peptic ulcer is the umbrella term for all types of gastrointestinal ulcers. Technically, an ulcer is at least 0.5 centimeters wide, but can be much larger [1] . Duodenal ulcers mainly occur in people between 20 and 50 years old, and gastric ulcers typically appear in people over age 40 [2] [3] . Duodenal ulcers are about four times more common than gastric ulcers (Nematollahi et al., 2011). Ulcers can be caused by varieties of factors which include: Helicobacter pylori infection, excessive gastric acid secretion, the use of Non Steroidal Anti-Inflammatory Drugs (NSAIDs), heavy use of alcohol, smoking and decreased mucosal defence against gastric acid [4] [5] . Lonchocarpus cyanescens is a species of shrub from Fabaceae family. Various studies have been carried out on the bioactivity, phythotherapeutic and anti-psychotic properties [6] - [12] of Lonchocarpus cy- anescens. Experimental studies on Lonchocarpus cyanescens triterpenoids showed that they can be used to treat ulcer [9] . Bioassay investigation carried out on the leaf extract of Lonchocarpus cyanescens triterpenoids for anti oxidant activities revealed that the anti oxidant properties correlated with its phenolic and flavonoid contents [11] . Molecular docking studies of the triterpenoids (Figure 1) using three (3) different malaria targets with PDB codes 3QS1, 1LS5 and 1SME were investigated using AutoDock vina. The docking studies showed that the ligands docked well with the targets [12] . Study on the molecular mechanism of anti ulcer action of Lonchocarpus cyanescens triterpenoids has not been reported. In this study, Lonchocarpus cyanescens triterpenoids are used as the ligands in molecular docking to validate its inhibitory activity of against ulcer.
2. Computational Procedures
The initial structures of proteins which are responsible for ulcer (1AFC, I AXM and 2AXM), were obtained in the form of pdb files from the Protein Data Bank [13] (www.pdb.org), and then to obtain the desired chains, multiple ligands and non-protein parts were deleted from the pdb files using Discovery Studio 4.1 visualizer. After that OpenBabel GUI version 2.3.2a and Spartan 14 version 1.18 were used to convert the pdb file format and optimize the geometry of the ligands, respectively. AutoDock Tools 1.5.6 and AutoDock Vina version 1.1.2
were used for molecular docking process [14] (http://autodock.scripps.edu) and to analyse the output of docking process, EduPymol version 1.7.4.4 was used. The exhaustiveness which determines how comprehensive the software search for the best binding mode was set to the default value of 8 which accompanied the ligands from the list of protein residues reported in the literature to characterise their binding sites for all the docking runs. Autodock tools (ADT) was used to add polar hydrogen to the prepared receptor. The three-dimensional affinity and electrostatic grid boxes were generated to cover the entire active site of the receptor. The number of grid points in x, y, z, was set to 40, 40, 40 with the spacing 1.000 Å. The grid centre (x, y and z) was varied from one receptor to another (see, Table 1). After the successful completion of the docking runs, different conformations of the ligands known as binding modes were obtained with their respective binding affinity and the stable one which happens to be the one with the lowest binding affinity was picked as the pose and was employed in the post-docking analysis using Edupymol by opening the receptors in the form of pdbqt file as well as the different binding modes of the ligands obtained after running AutoDock vina which were also in pdbqt file format to check the best binding mode that fitted well with the binding site cavity. The protein-ligand hydrogen interactions were also obtained in terms of atomic distance. The inhibition constants Ki were calculated using Equation (1)
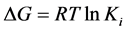
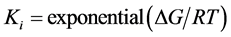 (1)
(1)
where ΔG is the binding affinity in kcal/mol, R is gas constant, 1.987 cal/mol/K and T is absolute temperature, assumed to be room temperature, 298.15 K [15] .
ADME and Drug Likeness Analysis
Molecular properties such as membrane permeability and bioavailability of leading compounds are always associated with some basic molecular descriptor such as logP (partition coefficient), molecular weight (mw), or number of hydrogen bond acceptors and donors in a molecule [16] . These molecular properties were used in formulating “rule of five” [17] . Lipinski’s rule states that molecule with good membrane permeability has MW ≤ 500, hydrogen bond donors ≤ 5 and hydrogen bond acceptor ≤ 10. Therefore, Lipinski’s Rule of Five was used to test the bioavailability characteristics such as Adsorption, Distribution, Metabolism and Elimination (ADME) of the ligands used in this study. In the present study, these molecular properties and druglikeness analysis were done using of Spartan 14 software.
![]()
Table 1. Grid centres (X, Y and Z) used for the docking runs.
3. Results and Discussion
Docking Score Results
From Table 2, the results of the studies revealed that the (the ligands used in this study) synthesized ligands compared favourably well with the standard ligands as shown in their binding affinities. The results also showed that OH derivative ligand has a better inhibitory effect (Figure 2(a), Figure 3(a) and Figure 4(a) for re-docking and Figure 2(b), Figure 2(c), Figure 3(b), Figure 3(c), Figure 4(b) and Figure 4(c) for OH and OCH3 respectively) than the OCH3 derivative which implies that OH derivative ligand will be more potent than OCH3 derivative in the treatment of ulcer. The OH and OCH3 derivatives triterpenoids gave binding affinities values (−7.2 Kcal/mol for 1AFC, −6.5 Kcal/mol for 1AXM and −6.2 Kcal/mol for 2AXM receptors) and (−6.7 Kcal/mol for 1AFC, −6.3 Kcal/ mol for 1AXM and −6.6 Kcal/mol for 2AXM receptors) respectively against the standard ligands value (−6.3 Kcal/mol for 1AFC, −6.2 Kcal/mol for 1AXM and −5.9 Kcal/mol for 2AXM receptors). Moreover, the smaller inhibition constant values (Ki) indicated that the ligands docked well to the receptor and that a relatively low concentration of the ligands is adequate to maximally occupy a ligand binding sites and trigger a physiological response. The binding affinity and inhibition constant for the OCH3 are higher than for OH. This is as a result of the inductive (electron releasing) effect of the methyl group. However, both derivatives have the capacity to inhibit ulcer, and as reported, this may be due to the high antioxidant properties of 14.83% at 0.50 mg/ml with scavenging effect of 63.89% and a strong correlation between antioxidant activity, phenol and flavonoid contents with correlation coefficient of 0.9906 and 0.9926 [18] . The findings from preliminary spectroscopic investigations that L. cyanescens extract contains essentially phyto-phenolic compounds was said to account for the use of the plants in the treatment of arthritic conditions and ulcers [6] [19] . Anti- ulcer & antioxidant activities of Hedranthera barteri [20] with possible involvement of H+, K+ ATPase inhibitory activity was assessed in cold-restraint (CRU), aspirin (ASP), alcohol (AL), pyloric ligation (PL) induced gastric ulcer models in rats and histamine-induced duodenal ulcer (HST) in guinea pigs. The effect of DMHBR (100 mg/kg) on gastric juice for free and total acidity, peptic activity and mucin secretion, using the pylorus ligated model, were evaluated. The H+, K+ and ATPase activity was assayed in gastric microsomes, spectrophotometri
![]()
Table 2. Docking score results for ulcer receptors.
Ei = Binding affinity (kcal/mol). Ki = Inhibition constant (µM).
cally. The in vitro anti-oxidant assays were explored through DPPH, nitric oxide, hydroxyl radical, superoxide anion scavenging assays. It was reported that DMHBR reduced the incidence of ulcers in CRU (63.3%), PL (58.5%), ASP (52.7%), HST (75.0%) and AL (53.87%). Also, reductions were observed in the free acidity (49.4%), total acidity (45.8%) and peptic activity (32.9%) with increase in the mucin secretion by 81.6%. DMHBR (60 - 100 μg/ml) inhibited the H+, K+ and ATPase activity with IC50 of 89.64 μg/ml compared with omeprazole
(10 - 50 μg/ml ) with IC50 of 32.26 μg/ml. DMHBR showed antioxidant activity with IC50 values of DPPH (397.69 μg/ml), nitric oxide (475.88 μg/ml), hydroxyl radical (244.22 μg/ml) and superoxide anion radical (285.20 μg/ml). It was observed that DMHBR showed anti-ulcer activity against experimentally-induced
peptic ulcer models and exhibited both cytoprotective and anti-secretory property. It exhibited a proton pump inhibition activity and its anti-ulcer properties was said to be partly ascribed to its antioxidant activities [20] . The results obtained from the molecular docking of the inhibitory activity of L. cyanescens active ligands follow the same pattern with the results obtained from molecular docking of L. cyanescens ligands as malaria inhibitors [12] .
The molecular descriptor (see Table 2) such as molecular weight, polar surface area volume, hydrogen bond donor, hydrogen bond acceptor, polarizability, lipophilicity and lipophobicity are vital to describe the bioactivity of a molecule. The Lipinski properties such as molecular weight (358.610 for OH and 372.637 for OCH3 ligand derivative), partition coefficient (3.20 for OH and 3.42 for OCH3 ligand derivative) which indicates the lipophilicity of the ligand (LogP), an estimate of a compound’s overall lipophilicity, it is a factor that influences the behaviour of a compound in a range of biological membranes, hepatetic clearance, selectivity and non-specific toxicity [21] . For good absorption, log P of a compound must be ≤5 therefore, these compounds are effective in terms of lipophilicity. Based on the Lipinski’s rule, the drug must have molecular weight value ≤ 500, Hydrogen Bond Donor ≤ 5, Hydrogen Bond Acceptor ≤ 10 and partition coefficient (LogP) value ≤ 5. From the results in Table 2, the ligands passed the Lipinski’s rule of five. Also, the Polar Surface Area (PSA) which is an indicator of the ligand hydrophilicity plays an important role in shaping the protein-ligand interaction by affecting the non-bonded contribution to the binding energy. Molecules with PSA greater than 140 Å2 are usually believe to be poor at penetrating cell membranes. For molecules to penetrate the blood-brain barrier, PSA should be less than 60 Å2 [16] [22] which is also fulfilled. The number of Hydrogen Bond Donor (1 for OH and 0 for OCH3 ligand derivative) and Hydrogen Bond Acceptor (1 for OH and 1 for OCH3 ligand derivative) obtained are also in good agreement with the Lipinski value for HBD and HBA (Table 2). The HOMO and LUMO are important properties that offer realistic and qualitative information about the excitation properties of molecules [23] [24] . The calculated HOMO are −6.53 eV for OH, and −6.18 eV for OCH3, and the calculated LUMO are −4.76 eV for OH, and −4.34 eV for OCH3 (Table 3). Therefore, the calculated band gaps are 1.77 eV for OH and 1.84 eV for OCH3. Band gap plays important role in the protein-ligand interaction, the lower the band gap, the easier the excitation and the better the ability of a molecule to donate electrons to the surrounding. The calculated ovality which measure the degree of deviation [24] of the cross section of the core from prefect circularity are 1.42 and 1.46 for OH and OCH3 derivatives, respectively. The calculated dipole moments are 1.18 and 0.92 for OH and OCH3, respectively. The dipole moment for OH derivative shows that the non-bonded interactions, the dipole-dipole interactions are essential in ligand-receptor interaction which has been shown to contribute about 3 to 5 kJ/mol [25] to the ligand-receptor energy.
Furthermore, Asn-18, Gln-127, Lys-118, Arg-122, Lys-113, Asn-114 residues are involved in the binding of the ligands to the 1AFC receptor. Similarly, Arg-122, Lys-118, Gln-127, Ala-129, Asn-114, Lys-128 residues are also involved in the binding of the ligands to 1AXM receptor while Arg-122, Lys-118, Leu-111, Lys-113, Gly-126, Arg-119 and Gln-127 residues are involved in the binding of the ligands to the 2AXM receptor binding cavity. The Hydrogen bond distance between the ligands and the proteins are shown in Table 4.
![]()
Table 3. Properties of the ligands used.
![]()
Table 4. Interaction between ligands and the receptors.
4. Conclusion
This study shows that Lonchocarpus cyanescens triterpenoids as the ligands in molecular docking to validate its inhibitory activity of against ulcer are effective and can be used to design drugs to treat ulcer because the results of the triterpenoid ligands are in good agreement with the standard ligands.