Enzyme-Mediated Enantioselective Hydrolysis of Aliphatic Dicarboxylic Acid Diesters ()
1. Introduction
The enzyme-mediated kinetic resolution of racemic alcohols and esters is one of the attractive methods for the preparation of optically active compounds [1] [2] [3] [4] . In our previous study, we succeeded in the enantioselective hydrolysis of poly(ethylene glycol) (PEG; av MW 4600)-supported carbonates (1) using porcine pancreas lipase (PPL; Scheme 1) [5] . In this case, two molecules of the optically active 1-phenylethanol (2) could be released from one molecule of the substrate 1, and the theoretical total yield of 2 was up to 200%. Unfortunately, the reactivity and enantioselectivitiy were moderate, and the amount of alcohols immobilized per gram of 1 (the loading capacity) was very low. This drawback is a limiting step for the preparative synthesis of the desirable enantiomer.
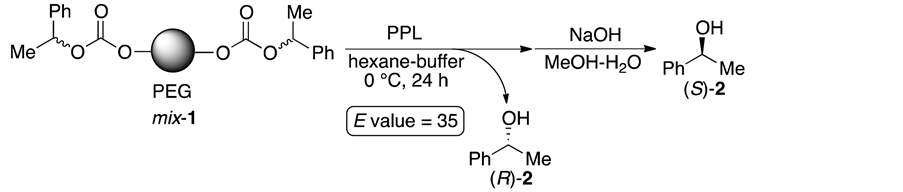
Scheme 1. Enzyme-mediated enantioselective hydrolysis of a PEG-supported substrate.
On the other hand, we also succeeded in the excellent enantioselective hydrolysis of aliphatic dicarboxylic acid monoesters 3 using lipase from Candida antarctica (Novozym 435; CAL-B), and the separation of the reaction products was achieved by a simple extraction procedure (Scheme 2) [6] . Then, we had noticed that the dicarboxylic acids would be a substitute for PEG spacer in Scheme 1, and the corresponding dicarboxylic acid diesters 4 could be a substrate for hydrolytic enzymes (Scheme 3). In this case, the gram-scale preparation of optically active compounds would be easy, because the molecular weight of the substrates would not be very high. Herein, we describe the enzyme-mediated enantioselective hydrolysis of aliphatic dicarboxylic acid diesters, and also report the methodical study of the substrate specificity. To the best of our knowledge, there have been only a very few reports on the enzyme-mediated enantioselective hydrolysis of diesters which release more than two equivalents of optically active alcohols [5] [7] .
2. Material and Methods
2.1. Materials
Novozym 435 (L4777, >5.0 U/mg) was obtained from Sigma-Aldrich Co. LLC. E. Merck Kieselgel 60 F254 Art.5715 was used for analytical TLC. Preparative TLC was performed on E. Merck Kieselgel 60 F254 Art.5744. Column chromatography was performed with Silica Gel 60N (63 - 210 mm, Kanto Chemical Co., Inc.). All other chemicals were also obtained from commercial sources.
2.2. Analytical Methods
1H (500 or 300 MHz) and 13C (125 or 75 MHz) NMR spectra were measured on a JEOL JNM-500 or AL-300, respectively, with tetramethylsilane (TMS) as the internal standard. IR spectra were recorded with Shimadzu IR Prestige-21 spectrometers. Mass spectra were obtained with a JEOL EI/FAB mate BU25 Instrument by the EI method. Optical rotations were measured with a Jasco DIP-1030 polarimeter. HPLC data were obtained on Shimadzu LC-10ADVP, SPD-10AVP, and μ7 Data Station (System Instruments Co., Ltd.) or Shimadzu LC-20AD, SPD-20A, and Smart Chrom (KYA technologies cooperation). GLC data were obtained on GL Sciences GC 353B, and μ7 Data Station (System Instruments Co., Ltd.).
2.3. Preparation of the Substrates for the Enzymatic Reaction
2.3.1. Mix-Bis(1-Phenylethyl) Glutarate (4b)
Under an argon atmosphere, 1-phenylethanol ((±)-2, 0.260 mL, 2.16 mmol) was added
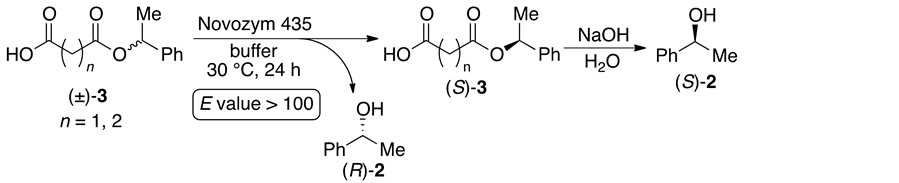
Scheme 2. Enzyme-mediated enantioselective hydrolysis of dicarboxylic monoesters.
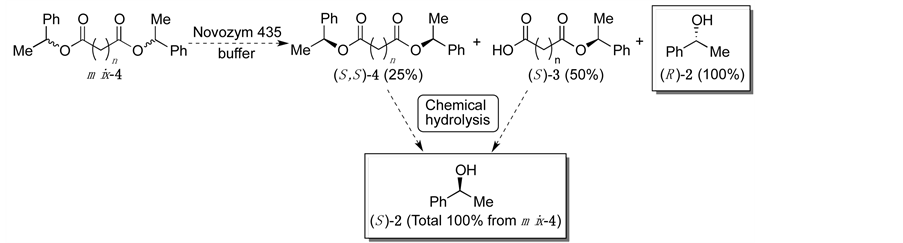
Scheme 3. Theoretical reaction of the enzyme-mediated enantioselective hydrolysis of racemic dicarboxylic acid diesters 4 (the numbers in parentheses are the theoretical % yields from mix-4).
to a solution of glutaric anhydride (500.7 mg, 4.388 mmol) in CH2Cl2 (10 mL). To the solution DMAP (1.047 g, 8.572 mmol) was added at 0˚C, and the mixture was stirred for 2 h at room temperature. After the mixture was washed with 2 M HCl, the products were extracted with CH2Cl2 (×3), and dried over Na2SO4. After evaporation in vacuo, the residue was purified by column chromatography on silica gel (hexane/AcOEt = 4/1) to give the (±)-4-((1-phenylethoxy)carbonyl)butanoic acid (3b) as a colorless oil (527.9 mg, 79%); IR (neat) 2980, 2936, 1732, 1709, 1495, 1452, 1375, 1287, 1246, 1207, 1155, 1063, 935, 762, 700 cm−1; 1H NMR (500 MHz, CDCl3) δ = 1.54 (d, J = 6.5 Hz, 3H), 1.95 (quintet, J = 7.5 Hz, 2H), 2.35 - 2.50 (m, 4H), 5.89 (q, J = 6.5 Hz, 1H), 7.25 - 7.41 (m, 5H); 13C NMR (125 MHz, CDCl3) δ = 19.8, 22.2, 32.9, 33.4, 72.5, 126.0, 127.9, 128.5, 141.5, 172.1, 179.0; MS m/z (EI, rel intensities) 236 (M+, 11%), 121 (100), 115 (77), 105 (100); HRMS m/z (EI) 236.1024 (calcd for C13H16O4: 236.1049, M+).
To a solution of (±)-3b (402.5 mg, 1.704 mmol) and (±)-2 (0.200 mL, 1.66 mmol) in CH2Cl2 (5 mL) were added DMAP (383.6 mg, 3.140 mmol) and DCC (658.3 mg, 3.190 mmol) at 0˚C, and the mixture was stirred overnight at room temperature. After the mixture was filtered through a celite pad using CH2Cl2, the filtrate was washed with 0.5 M HCl (x2), and the organic layer was dried over Na2SO4. After evaporation in vacuo, the residue was purified by column chromatography on silica gel (hexane/AcOEt = 8/1) to give mix-4b as a colorless oil (434.5 mg, 75%); IR (neat) 2978, 2931, 2360, 1734, 1450, 1375, 1250, 1173, 1063, 762, 698 cm−1; 1H NMR (300 MHz, CDCl3) δ = 1.52 (d, J = 6.5 Hz, 6H), 1.94 (quintet, J = 7.5 Hz, 2H), 2.37 (t, J = 7.5 Hz, 4H), 5.88 (q, J = 6.5 Hz, 2H), 7.21 - 7.38 (m, 10H); 13C NMR (125 MHz, CDCl3) δ = 20.1, 22.3, 33.5, 72.3, 126.0, 127.9, 128.5, 141.6, 172.2; MS m/z (EI, rel intensities) 341 (M+, 6.5%), 235 (100), 120 (100), 105 (100); HRMS m/z (EI) 341.1754 (calcd for C21H25O4: 341.1753, M+ + H).
The compound 4b is a 1:1 mixture of dl- and meso-form diastereomers. The ratio was determined by HPLC analysis using CHIRALCEL AS-H (Daicel Chemical Industries, Ltd.): eluent, hexane/2-propanol = 95/5; flow rate, 0.5 mL/min; 254 nm; temperature, 25˚C; retention time, 12.2 (R, R) and12.8 [meso and (S, S)] min.
Other substrates 4a, 18b, 19b, 20b, 21b, 22b and 23b were synthesized by the same procedure.
2.3.2. Mix-Bis(1-Phenylethyl) Succinate (4a)
Yield 73% from (±)-2 in 2 steps (a colorless oil); IR (neat) 2980, 2931, 2363, 1734, 1456, 1450, 1375, 1207, 1159, 1062, 862, 761, 698 cm−1; 1H NMR (500 MHz, CDCl3) δ = 1.51 (d, J = 7.0 Hz) and 1.52 (d, J = 6.5 Hz) (6H), 2.57 - 2.75 (m, 4H), 5.89 (q, J = 6.5 Hz, 2H), 7.25 - 7.38 (m, 10H); 13C NMR (75 MHz, CDCl3) δ = 22.1, 22.2, 29.4, 29.5, 72.7, 126.0, 126.1, 127.9, 128.5, 141.5, 171.4; MS m/z (EI, rel intensities) 354 (M+, 0.8%), 221 (100), 149 (100), 121 (100), 105 (100); HRMS m/z (EI) 326.1510 (calcd for C20H22O4: 326.1518, M+).
2.3.3. Mix-Bis(1-Phenylpropyl) Glutarate (18b)
Yield 90% from (±)-6 in 2 steps (a colorless oil); IR (neat) 2972, 2936, 2876, 1734, 1456, 1381, 1246, 1171, 1084, 964, 756, 700 cm−1; 1H NMR (300 MHz, CDCl3) δ = 0.87 (t, J = 7.5 Hz, 6H), 1.65 - 2.02 (m, 6H), 2.30 - 2.46 (m, 4H), 5.66 (t, J = 7.0 Hz, 2H), 7.20 - 7.41 (m, 10H); 13C NMR (125 MHz, CDCl3) δ = 9.9, 20.2, 29.3, 33.5, 77.4, 126.5, 127.8, 128.4, 140.5, 172.2; MS m/z (EI, rel intensities) 369 (M+ + H, 1.5%,), 368 (M+, 0.5), 249 (100), 233 (4.6), 135 (100), 119 (100); HRMS m/z (EI) 368.2013 (calcd for C23H28O4: 382.2144, M+).
2.3.4. Mix-Bis(1-Phenylpropan-2-yl) Glutarate (19b)
Yield 89% from (±)-7 in 2 steps (a colorless oil); IR (neat) 2976, 2932, 1719, 1491, 1452, 1377, 1251, 1177, 1134, 1059, 746, 700 cm−1; 1H NMR (500 MHz, CDCl3) δ = 1.215 (d, J = 6.5 Hz) and 1.218 (d, J = 6.5 Hz) (6H), 1.81 (tt, J1 = J2 = 7.5 Hz, 2H), 2.18 - 2.25 (m, 4H), 2.75 (dd, J1 = 6.5 Hz, J2 = 13.5 Hz, 2H), 2.90 (ddd, J1 = 1.5 Hz, J2 = 6.5 Hz, J3 = 13.5 Hz 2H), 5.12 (tq, J1 = J2 = 6.5 Hz, 2H), 7.16 - 7.30 (m, 10H); 13C NMR (125 MHz, CDCl3) δ = 19.7, 20.2, 33.6, 42.4, 71.6, 126.6, 128.4, 129.5, 137.7, 172.5; MS m/z (EI, rel intensities) 369 (M+ + H, 29%), 368 (M+, 1.0), 251 (100), 233 (100), 135 (79), 119 (100); HRMS m/z (EI) 368.1946 (calcd for C23H28O4: 368.1988, M+).
2.3.5. Mix-Bis(1-(Naphthalen-2-yl)Ethyl) Glutarate (20b)
Yield 53% from (±)-8 in 2 steps (a colorless solid); IR (KBr) 2927, 1734, 1236, 1171, 1066, 908, 777, 733 cm−1; 1H NMR (300 MHz, CDCl3) δ = 1.679 (d, J = 6.5 Hz) and 1.683 (d, J = 6.5 Hz) (6H), 1.92 - 2.06 (m, 2H), 2.33 - 2.52 (m, 4H), 6.64 (q, J = 6.5 Hz, 2H), 7.36 - 7.60 (m, 8H), 7.78 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 7.0 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ = 20.2, 21.7, 33.6, 69.5, 123.1, 125.4, 125.7, 126.3, 128.4, 128.9, 130.2, 133.8, 137.4, 172.2; MS m/z (EI, rel intensities) 440 (M+, 23%), 285 (1.2), 171 (100), 155 (100), 115 (27); HRMS m/z (EI) 440.1990 (calcd for C29H28O4: 440.1988, M+).
2.3.6. Mix-Bis(1-(Naphthalen-1-yl)Ethyl) Glutarate (21b)
Yield 91% from (±)-9 in 2 steps (a colorless oil); IR (neat) 2980, 2931, 1722, 1373, 1277, 1171, 1049, 824, 746 cm−1; 1H NMR (300 MHz, CDCl3) δ = 1.603 (d, J = 6.5 Hz) and 1.609 (d, J = 6.5 Hz) (6H), 1.91 - 2.07 (m, 2H), 2.41 (t, J = 7.0 Hz, 4H), 6.05 (dq, J1 = 1.5 Hz, J2 = 6.5 Hz, 2H), 7.39 - 7.55 (m, 6H), 7.75 - 7.90 (m, 8H); 13C NMR (125 MHz, CDCl3) δ = 20.2, 21.7, 33.6, 69.5, 123.1, 125.4, 125.7, 126.3, 128.4, 128.9, 130.2, 133.8, 137.4, 172.2; MS m/z (EI, rel intensities) 440 (M+, 23%), 285 (1.2), 171 (100), 155 (100), 115 (27); HRMS m/z (EI) 440.1983 (calcd for C29H28O4: 440.1988, M+).
2.3.7. Mix-Bis(4-Phenylbutan-2-yl) Glutarate (22b)
Yield 27% from (±)-10 in 2 steps (a colorless oil); IR (neat) 2974, 2932, 2361, 2344, 1732, 1454, 1377, 1250, 1179, 1130, 1051, 748, 698 cm−1; 1H NMR (300 MHz, CDCl3) δ = 1.25 (d, J = 6.0 Hz, 6H), 1.71 - 2.05 (m, 4H), 2.35 (t, J = 7.5 Hz, 4H), 2.50 - 2.77 (m, 4H), 4.95 (tq, J1 = J2 = 6.0 Hz, 2H), 7.09 - 7.22 (m, 6H), 7.22 - 7.35 (m, 4H); 13C NMR (125 MHz, CDCl3) δ = 20.2, 29.8, 31.9, 37.6, 70.6 and 70.8, 125.9 and 126.0, 128.4, 128.5, 141.6, 172.7; MS m/z (EI, rel intensities) 396 (M+, 100%), 291 (4.8), 273 (81), 265 (18), 247 (57), 177 (38), 149 (64), 133 (100), 115 (100), 105 (100); HRMS m/z (EI) 396.2301 (calcd for C25H32O4: 396.2301, M+).
2.3.8. Mix-Bis(4-(Benzyloxy)Butan-2-yl) Glutarate (23b)
Yield 40% from (±)-11 in 2 steps (a colorless oil); IR (neat) 2976, 2934, 2862, 1728, 1454, 1377, 1252, 1201, 1028, 739, 698 cm−1; 1H NMR (500 MHz, CDCl3) δ = 1.23 (d, J = 6.0 Hz, 6H), 1.72 - 2.00 (m, 6H), 2.27 (t, J = 7.5 Hz, 2H), 3.43 - 3.53 (m, 4H), 4.466 (d, J = 12.0 Hz, 2H), 4.471 (d, J = 12.0 Hz, 2H), 5.04 - 5.14 (m, 2H), 7.24 - 7.38 (m, 10H); 13C NMR (125 MHz, CDCl3) δ = 20.4, 33.7, 36.1, 66.6, 68.6, 73.1, 127.7, 127.8, 128.5, 138.4, 172.6; MS m/z (EI, rel intensities) 456 (M+, 6%), 365 (74), 349 (42), 277 (100), 179, (85), 163 (100), 121 (100), 114 (100), 107 (100); HRMS m/z (EI) 456.2509 (calcd for C27H36O4: 456.2512, M+).
2.3.9. Mix-Bis(1-Phenylethyl) Adipate (4c)
To a solution of adipic acid (1.00 g, 6.84 mmol) in CH2Cl2 (3 mL) were added (±)-2 (2.50 mL, 20.8 mmol), a solution of DMAP (1.67 g, 13.7 mmol) in CH2Cl2 (9 mL) and a solution of DCC (3.53 g, 17.1 mmol) in CH2Cl2 (9 mL) at 0˚C, and the mixture was stirred overnight at room temperature. After the mixture was filtered through a celite pad using CH2Cl2, the filtrate was washed with 0.5 M HCl (x2), and the organic layer was dried over Na2SO4. After evaporation in vacuo, the residue was purified by column chromatography on silica gel (hexane/AcOEt = 4/1) to give mix-4c as a colorless oil (2.20 g, 90%); IR (neat) 2978, 2931, 2361, 1734, 1452, 1375, 1244, 1170, 1064, 1029, 761, 698 cm−1; 1H NMR (300 MHz, CDCl3) δ = 1.51 (d, J = 6.5 Hz, 6H), 1.56 - 1.74 (m, 4H), 2.22 - 2.42 (m, 4H), 5.86 (q, J = 6.5 Hz, 2H), 7.21 - 7.39 (m, 10H); 13C NMR (75 MHz, CDCl3) δ = 22.2, 24.3, 34.2, 72.5, 126.0, 127.8, 128.5, 141.7, 172.5; MS m/z (EI, rel intensities) 354 (M+, 1.0%), 249 (100), 233 (17), 121 (100); HRMS m/z (EI) 354.1816 (calcd for C22H26O4: 354.1831, M+).
The compound 4c is a 1:1 mixture of dl- and meso-form diastereomers. The ratio was determined by HPLC analysis using CHIRALCEL OD-H (Daicel Chemical Industries, Ltd.): eluent, hexane/2-propanol = 95/5; flow rate, 0.5 mL/min; 254 nm; temperature, 25˚C; retention time, 17.0 (R, R), 18.4 (meso), and 19.8 (S, S) min.
Other substrates 4d-f, 18c, 19c, 20c, 21c, 22c and 23c were synthesized by the same procedure.
2.3.10. Mix-Bis(1-Phenylethyl) Heptanedioate (4d)
Yield 58% from pimelic acid (a colorless oil); IR (neat) 2978, 2933, 2363, 1734, 1452, 1373, 1250, 1172, 1065, 1030, 762, 698 cm−1; 1H NMR (500 MHz, CDCl3) δ = 1.24 - 1.33 (m, 2H), 1.52 (d, J = 6.5 Hz, 6H), 1.58 - 1.67 (m, 4H), 2.30 (t, J = 6.5 Hz, 2H), 2.31 (t, J = 7.5 Hz, 2H), 5.88 (q, J = 6.5 Hz, 2H), 7.24 - 7.38 (m, 10H); 13C NMR (75 MHz, CDCl3) δ = 22.2, 24.5, 28.5, 34.3, 72.1, 126.0, 127.8, 128.5, 141.7, 172.8; MS m/z (EI, rel intensities) 369 (M+ + H, 1.1%), 368 (M+, 1.0), 263 (100), 149 (5.5), 143 (100), 121 (100); HRMS m/z (EI) 369.2085 (calcd for C23H29O4: 369.2066, M+ + H).
2.3.11. Mix-Bis(1-Phenylethyl) Octanedioate (4e)
Yield 84% from suberic acid (a colorless oil); IR (neat) 2978, 2934, 2361, 1734, 1450, 1248, 1171, 1065, 1030, 762, 698 cm−1; 1H NMR (500 MHz, CDCl3) δ = 1.24 - 1.33 (m, 4H), 1.53 (d, J = 6.5 Hz, 6H), 1.55 - 1.64 (m, 4H), 2.297 (t, J = 7.5 Hz, 2H), 2.302 (t, J = 7.5 Hz, 2H), 5.88 (q, J = 6.5 Hz, 2H), 7.24 - 7.31 (m, 2H), 7.31 - 7.39 (m, 8H); 13C NMR (75 MHz, CDCl3) δ = 22.2, 24.7, 28.6, 34.4, 72.0, 126.0, 127.8, 128.4, 141.8, 172.9; MS m/z (EI, rel intensities) 383 (M+ + H, 1.5%), 277 (100), 157 (100), 139 (91), 121 (100), 105 (100); HRMS m/z (EI) 383.2220 (calcd for C24H31O4: 383.2222, M+ + H).
2.3.12. Mix-Bis(1-Phenylethyl) Decanedioate (4f)
Yield 74% from sebacic acid (a colorless oil); IR (neat) 2930, 2855, 2361, 1734, 1452, 1373, 1244, 1170, 1065, 1030, 762, 698 cm−1; 1H NMR (300 MHz, CDCl3) δ = 1.18 - 1.34 (m, 8H), 1.52 (d, J = 7.0 Hz, 6H), 1.52 - 1.68 (m, 4H), 2.31 (t, J = 7.5 Hz, 3H), 5.88 (q, J = 6.5 Hz, 2H), 7.23 - 7.38 (m, 10H); 13C NMR (75 MHz, CDCl3) δ = 22.2, 24.9, 28.97, 29.01, 34.6, 72.0, 126.0, 127.8, 128.4, 141.8, 173.1; MS m/z (EI, rel intensities) 411 (M+ + H, 0.9%), 305 (100), 287 (12), 185 (100), 139 (100), 121 (100), 105 (100); HRMS m/z (EI) 411.2531 (calcd for C26H35O4: 411.2535, M+ + H).
2.3.13. Mix-Bis(1-Phenylpropyl) Adipate (18c)
Yield 84% from adipic acid (a colorless oil); IR (neat) 2968, 2936, 2876, 1732, 1494, 1454, 1381, 1240, 1168, 1084, 968, 912, 756, 700 cm−1; 1H NMR (300 MHz, CDCl3) δ = 0.87 (t, J = 7.5 Hz, 6H), 1.56 - 1.58 (m, 4H), 1.59 - 2.00 (m, 4H), 2.25 - 2.43 (m, 4H), 5.65 (t, J = 7.0 Hz, 2H), 7.22 - 7.41 (m, 10H); 13C NMR (125 MHz, CDCl3) δ = 9.9, 24.4, 29.3, 34.1, 126.5, 127.8, 128.4, 140.6, 172.6; MS m/z (EI, rel intensities) 382 (M+, 1.0), 263 (100), 235 (100), 135 (100), 129 (100), 119 (100); HRMS m/z (EI) 382.2107 (calcd for C24H30O4: 382.2144, M+).
2.3.14. Mix-Bis(1-Phenylpropan-2-yl) Adipate (19c)
Yield 72% from adipic acid (a colorless oil); IR (neat) 2976, 2932, 2369, 1732, 1452, 1375, 1248, 1175, 1132, 1076, 1059, 746, 700 cm−1; 1H NMR (500 MHz, CDCl3) δ = 1.21 (d, J = 6.0 Hz, 6H), 1.44 - 1.55 (m, 4H), 2.16 - 2.25 (m, 4H), 2.75 (dd, J1 = 6.5 Hz, J2 = 13.5 Hz, 2H), 2.90 (dd, J1 = 7.0 Hz, J2 = 13.5 Hz, 2H), 5.12 (qt, J1 = J2 = 6.5 Hz, 2H), 7.15 - 7.31 (m, 10H); 13C NMR (125 MHz, CDCl3) δ = 19.7, 24.4, 34.3, 42.4, 71.5, 126.6, 128.4, 129.5, 137.7, 172.9; MS m/z (EI, rel intensities) 383 (M+ + H, 13%), 382 (M+, 1.1), 265 (100), 247 (100), 135 (18), 129 (100), 119 (100); HRMS m/z (EI) 382.2099 (calcd for C24H30O4: 382.2144, M+).
2.3.15. Mix-Bis(2-(Naphthalen-2-yl)Ethyl) Adipate (20c)
Yield 51% from adipic acid (a colorless solid); IR (KBr) 2934, 1721, 1510, 1375, 1256, 1167, 1053, 804, 781 cm−1; 1H NMR (300 MHz, CDCl3) δ = 1.68 (d, J = 6.5 Hz, 2H), 1.82 - 2.00 (m, 4H), 2.30 - 2.44 (m, 4H), 6.64 (q, J = 6.5 Hz, 2H), 7.38 - 7.55 (m, 6H), 7.56 (d, J = 7.5 Hz, 1H), 7.78 (d, J = 8.5 Hz, 1H), 7.86 (d, J = 7.5 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ = 21.8, 24.5, 34.3, 69.5, 123.3, 125.4, 125.8, 126.4, 128.5, 129.0, 130.3, 133.9, 137.5, 172.7; MS m/z (EI, rel intensities) 454 (M+, 43%), 299 (1.3), 171 (100), 155 (100), 129 (64); HRMS m/z (EI) 454.2144 (calcd for C30H30O4: 454.2144, M+).
2.3.16. Mix-Bis(1-(Naphthalen-1-yl)Ethyl) Adipate (21c)
Yield 50% from adipic acid (a colorless solid); IR (neat) 2986, 2928, 1728, 1375, 1227, 1188, 1057, 822, 748 cm−1; 1H NMR (300 MHz, CDCl3) δ = 1.59 (d, J = 6.5 Hz, 6H), 1.62 - 1.72 (m, 4H), 2.30 - 2.42 (m, 4H), 6.03 (q, J = 6.5 Hz, 2H), 7.41 - 7.51 (m, 6H), 7.75 - 7.87 (m, 8H); 13C NMR (125 MHz, CDCl3) δ = 21.8, 24.5, 34.3, 69.5, 123.3, 125.4, 125.8, 126.4, 128.5, 129.0, 130.3, 133.9, 137.5, 172.7; MS m/z (EI, rel intensities) 454 (M+, 40%), 299 (2.7), 171 (100), 155 (100), 129 (48); HRMS m/z (EI) 454.2146 (calcd for C30H30O4: 454.2144, M+).
2.3.17. Mix-Bis(4-Phenylbutan-2-yl) Adipate (22c)
Yield 69% from adipic acid (a colorless oil); IR (neat) 2974, 2934, 2864, 2363, 0730, 1495, 1454, 1377, 1244, 1177, 1130, 1051, 748, 700 cm−1; 1H NMR (500 MHz, CDCl3) δ = 1.24 (d, J = 6.5 Hz, 6H), 1.62 - 1.73 (m, 4H), 1.75 - 1.84 (m, 2H), 1.87 - 1.97 (m, 2H), 2.26 - 2.36 (m, 4H), 2.55 - 2.70 (m, 4H), 4.94 (tq, J1 = J2 = 6.0 Hz, 2H), 7.14 - 7.21 (m, 6H), 7.24 - 7.31 (m, 4H); 13C NMR (125 MHz, CDCl3) δ = 20.1, 24.5, 31.8, 34.3, 70.4, 125.9, 128.3, 128.4, 141.5, 173.0; MS m/z (EI, rel intensities) 410 (M+, 11.6%), 273 (9.2), 235 (21), 177 (8.4), 164 (6.5), 149 (12), 133 (100), 106 (100); HRMS m/z (EI) 410.2457 (calcd for C26H34O4: 410.2457, M+).
2.3.18. Mix-Bis(4-(Benzyloxy)Butan-2-yl) Adipate (23c)
Yield 51% from adipic acid (a colorless oil); IR (neat) 2976, 2934, 2864, 1730, 1497, 1453, 1377, 1246, 1180, 1099, 912, 745, 698 cm−1; 1H NMR (500 MHz, CDCl3) δ = 1.23 (d, J = 6.5 Hz, 6H), 1.55 - 1.65 (m, 4H), 1.76 - 1.93 (m, 4H), 2.19 - 2.28 (m, 4H), 3.44 - 3.53 (m, 4H), 4.468 (d, J = 12.0 Hz, 2H), 4.472 (d, J = 12.0 Hz, 2H), 5.03 - 5.12 (m, 2H), 7.24 - 7.38 (m, 10H); 13C NMR (125 MHz, CDCl3) δ = 20.4, 24.5, 34.3, 36.1, 66.7, 68.5, 68.8, 73.1, 127.7, 127.8, 128.5, 138.4, 172.9; MS m/z (EI, rel intensities) 470 (M+, 2.8%), 379 (38), 363 (13), 273 (100), 201 (100), 183 (100), 162 (100), 108 (100); HRMS m/z (EI) 470.2705 (calcd for C28H38O6: 470.2668, M+).
2.4. Enzymatic Hydrolysis of Mix-4c with Novozym 435
2.4.1. Typical Procedure
To a 200-mL Erlenmeyer flask containing 142 mg of mix-4c (0.401 mmol) was added 40 mL of 0.1 M phosphate buffer (pH 6.5). To the mixture was added 40 mg of Novozym 435 (Sigma L4777, >5.0 U/mg), and the flask was shaken at 120 min−1 for 24 h at 30˚C. After addition of 2 M HCl to the mixture, the products were extracted with Et2O (x3), and the organic layer was washed with brine and dried over Na2SO4. After the organic phase was evaporated in vacuo, the residue was purified by flash column chromatography on silica gel (hexane/AcOEt = 7/1-3/1) to give (S, S)-4c (34.1 mg, 25%, >99% ee), (S)-3c (47.4 mg, 50%, >99% ee), and (R)-2 (47.0 mg, 96%, >99% ee). The ee of (R)-2 was determined by GC analysis with a chiral column. The remaining (S, S)-4c and (S)-3c were chemically hydrolyzed with 2 M NaOH in MeOH and in H2O, respectively, to afford the corresponding alcohol (S)-2. The ee of the resulting (S)-2 was regarded as the ee of the original ester.
GC conditions: column, CP-Cyclodextrin-B-236-M19 (Agilent Technologies, Inc.), 0.25 mm × 50 m; injection, 140˚C; detection, 140˚C; oven, 120˚C; carrier gas, He; head pressure, 2.4 kg/cm2; retention time, 14.6 (R) and 15.2 (S) min.
2.4.2. Preparative Scale Procedure
To 3000-mL Erlenmeyer flask containing 1.43 g of mix-4c (4.00 mmol) was added 400 mL of 0.1 M phosphate buffer (pH 6.5). To the mixture was added 400 mg of Novozym 435, and the flask was shaken at 120 min−1 for 24 h at 30˚C. After addition of 2 M HCl to the mixture, the products were extracted with Et2O (x3), and the organic layer was washed with brine and dried over Na2SO4. After the organic phase was evaporated in vacuo, the residue was purified by flash column chromatography on silica gel (hexane/Et2O = 6/1) to give (S, S)-4c (330 mg, 23%), (S)-3c (492 mg, 49%), and (R)-2 (484 mg, 99%, >99% ee). All the spectral data (1H and 13C NMR, IR, and MS) of 4c and 2 were in full agreement with those of the racemate 4c and the commercial source 2, respectively.
(S, S)-4c: [α]D26 = −86.9 (c1.29, MeOH).
(S)-3c: IR (neat) 2955, 1736, 1709, 1495, 1452, 1375, 1287, 1175, 1065, 762, 700 cm−1; 1H NMR (500 MHz, CDCl3) δ = 1.53 (d, J = 6.5 Hz, 3H), 1.60 - 1.73 (m, 4H), 2.29 - 2.42 (m, 4H), 5.89 (q, J = 6.5 Hz, 1H), 7.25 - 7.38 (m, 5H); 13C NMR (125 MHz, CDCl3) δ = 22.3, 24.1, 24.4, 33.7, 34.3, 72.4, 126.2, 128.0, 128.6, 141.7, 172.7, 179.6; MS m/z (EI, rel intensities) 250 (M+, 66%), 222 (91), 129 (100), 121 (100); HRMS m/z (EI) 250.1200 (calcd for C14H18O4: 250.1205, M+); [α]D25 = −61.6 (c1.08, MeOH).
(R)-2: [α]D24 = +38.4 (c1.04, MeOH) (>99% ee); lit. [α]D20 = +45 (c 5.15, MeOH) [8] .
To the diester (S, S)-4c in MeOH (5 mL) was added 2 M NaOH (2 mL), and the mixture was stirred at rt. The products were extracted with Et2O (x3), and the organic layer was washed with brine and dried over Na2SO4. After the organic phase was evaporated in vacuo, the residue was purified by flash column chromatography on silica gel (hexane/AcOEt = 2/1) to give (S)-2 (213 mg, 188%, >99% ee).
To the monoester (S)-3c was added 2 M NaOH (5 mL), and the mixture was stirred at rt. The products were extracted with Et2O (x3), and the organic layer was washed with brine and dried over Na2SO4. After the organic phase was evaporated in vacuo, the residue was purified by flash column chromatography on silica gel (hexane/AcOEt = 2/1) to give (S)-2 (215 mg, 90%, >99% ee).
(S)-2: [α]D28 = ?43.0 (c 1.13, MeOH) (>99% ee).
2.5. Data for the Alcohols Derived from the Enzymatic Hydrolysis of Mix-Dicarboxylic Acid Diesters
The reactions of the other substrates were carried out by the same procedure. The results were shown in the text. All the spectral data (1H and 13C NMR, IR, and MS) were in full agreement with those of the racemates, commercial sources, or those reported.
1-phenylpropan-1-ol (6)
(S)-6: [α]D23 = −44.5 (c 1.26, CHCl3) (>99% ee); lit. [α]D20 = −47.0 (c 1.00, CHCl3) [9] .
(R)-6: [α]D24 = +44.2 (c 1.83, CHCl3) (>99% ee).
GC conditions: column, CP-Cyclodextrin-B-236-M19 (Chrompack), 0.25 mm × 50 m; injection, 140˚C; detection, 140˚C; oven, 120˚C; carrier gas, He; head pressure, 2.4 kg/cm2; retention time, 22.6 (R) and 23.4 (S) min.
1-phenylpropan-2-ol (7)
(S)-7: [α]D26 = −32.6 (c1.67, CHCl3) (>99% ee).
(R)-7: [α]D26 = +40.3 (c 0.49, CHCl3) (>99% ee); lit. [α]D20 = −37.6 (c 5.00, CHCl3) [10] .
GC conditions: column, CP-Cyclodextrin-B-236-M19 (Chrompack), 0.25 mm × 50 m; injection, 130˚C; detection, 130˚C; oven, 110˚C; carrier gas, He; head pressure, 2.4 kg/cm2; retention time, 27.1 (R) and 27.5 (S) min.
1-(2-naphthyl)ethanol (8)
(S)-8: [α]D24 = −36.2 (c 0.86, MeOH) (>99% ee).
(R)-8: [α]D25 = +37.4 (c1.10, MeOH) (>99% ee); lit. [α]D = +34.6 (c 1.20, MeOH) [11] .
GC conditions: column, CP-Cyclodextrin-B-236-M19 (Chrompack), 0.25 mm × 50 m; injection, 180˚C; detection, 180˚C; oven, 160˚C; carrier gas, He; head pressure, 2.4 kg/cm2; retention time, 41.9 (R) and 42.9 (S) min.
1-(1-naphthyl)ethanol (9)
(S)-8: [α]D23 = −13.2 (c 0.84, MeOH) (49% ee).
(R)-8: [α]D24 = +22.3 (c 0.61, MeOH) (75% ee); lit. [α]D25 = +45.0 (c 2.00, MeOH) [12] .
GC conditions: column, CP-Cyclodextrin-B-236-M19 (Chrompack), 0.25 mm × 50 m; injection, 180˚C; detection, 180˚C; oven, 16˚C; carrier gas, He; head pressure, 2.4 kg/cm2; retention time, 43.0 (R) and 44.3 (S) min.
4-phenylbutan-2-ol (10)
(S)-10: [α]D27 = +11.9 (c1.04, CHCl3) (98% ee).
(R)-11: [α]D27 = −13.1 (c0.83, CHCl3) (81% ee); lit. [α]D21 = −14.0 (c 1.63, CHCl3) [13] .
HPLC conditions: column, CHIRALCEL OD-H (Daicel Chemical Industries, Ltd.); eluent, hexane/2-propanol = 90/10; flow rate, 0.5 mL/min; 254 nm; temperature, 25˚C; retention time, 12.6 (R) and 16.3 (S) min.
4-benzyloxybutan-2-ol (11)
(S)-11: [α]D24 = +14.5 (c1.34, MeOH) (99% ee); lit. [α]D27 = +19.0 (c 0.95, MeOH) [14] .
(R)-11: [α]D25 = −12.3 (c1.05, MeOH) (91% ee).
HPLC conditions: column, CHIRALCEL OD-H (Daicel Chemical Industries, Ltd.); eluent, hexane/2-propanol = 90/10; flow rate, 0.5 mL/min; 254 nm; temperature, 25˚C; retention time, 12.8 (S) and 14.0 (R) min.
2.6. Enzymatic Hydrolysis of (±)-Acetates Derived from Alcohols with Novozym 435
Racemic acetates were prepared from the corresponding alcohols by a usual method using acetic anhydride in pyridine.
Enzymatic reactions of the acetates were carried out using 20 mM of the substrates with Novozyme 435 (40 mg) in 0.1 M phosphate buffer (pH 6.5, 40 mL) at 30˚C for 24 h. The analytical methods of the products were almost same as those in the case of the dicarboxylic acid diesters mentioned above.
3. Results and Discussion
3.1. Concept of the Enzymatic Hydrolysis of Dicarboxylic Acid Diesters
Based on our concept of the enzymatic hydrolysis of mix-4, the process involving the production of (R)-2 contains two different steps, which are the first hydrolysis of the diesters mix-4 and the following second hydrolysis of the resulting monoester 3 (Scheme 3). When the reactions of the diesters mix-4, which contains the racemates ((S, S)-4 and (R, R)-4) and meso-4 in the ratio 1:1, theoretically proceed, the yield of the resulting alcohol (R)-2 could be 100%. In a similar way, the unreactive (S, S)-4 and (S)-3 could be obtained in 25% and 50% yields, respectively, and the following chemical hydrolysis of esters 4 and 3 could also give (S)-2 in 100% total yield based on the amount of mix-4. According to our previous study [6] , the highly enantioselective hydrolysis of monoesters (±)-3 using Novozym 435 should be expected. We then specifically focused on the reactivity of the diesters 4.
3.2. Preparation of Racemic Dicarboxylic Acid Diesters as the Substrate
For the synthesis of the substrates, the racemate (±)-2 was combined with succinic anhydride or glutaric anhydride using DMAP in CH2Cl2 to give the corresponding (±)-3a (n = 2) and 3b (n = 3), respectively (Scheme 4). The monoesters were coupled with another (±)-2 using DCC and DMAP in CH2Cl2 to afford the substrates mix-4a and 4b, respectively. On the other hand, the diesters mix-4c (n = 4), 4d (n = 5), 4e (n = 6) and 4f (n = 8) were prepared by the direct coupling of (±)-2 with adipic acid, pimelic acid, suberic acid, and sebacic acid, respectively. The other substrates were synthesized by the same procedure (Scheme 5). HPLC analyses of mix-4b and 4c showed that the compounds were almost 1:1 mixtures of the diastereomers, and we then decided that the diastereomeric ratios of the prepared diesters 4 should be 1:1, regardless of the synthetic process.
3.3. Enzymatic Hydrolysis of Racemic Dicarboxylic Diester mix-4
We initially took notice of the carbon number between the two ester parts of the substrates, and the enzymatic reactions using Novozym 435 of several diesters mix-4a-f were carried out. After the enzymatic reactions of mix-4 (0.4 mmol) using Novozym 435 (80 mg) in 0.1 M phosphate buffer (pH 6.5, 40 mL), the isolated yields of the compounds were determined after purification. The remaining diesters and monoesters were
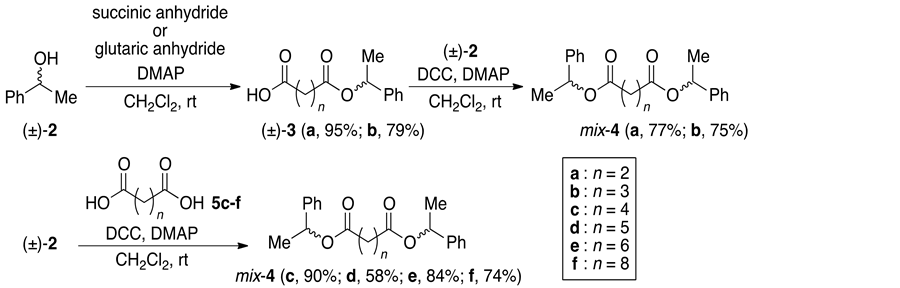
Scheme 4. Synthesis of the substrates mix-4.
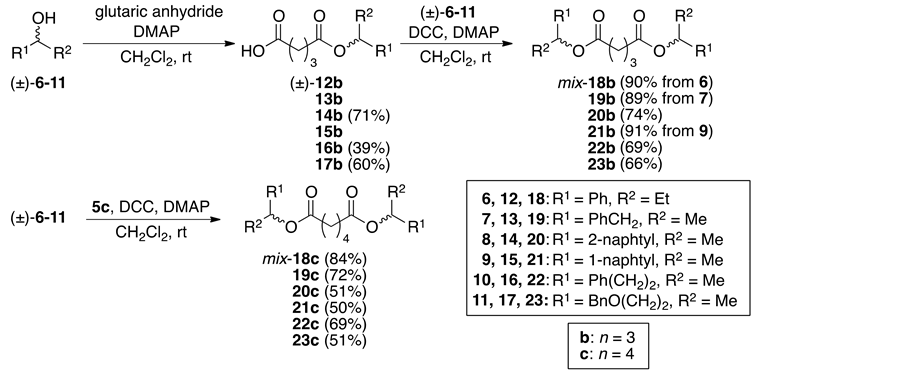
Scheme 5. Synthesis of the substrates mix-18b-23b and 18c-23c.
sequentially hydrolyzed with NaOH. The enantiomeric excesses (ee) of the resulting 2 were evaluated by a chiral GLC analysis, and the results are summarized in Table 1. Surprisingly, in all cases, the enzymatic hydrolyses of 4 proceeded with excellent enantioselectivities to afford the corresponding optically active compounds. Furthermore, the excellent ee values of the resulting (R)-2 from the enzymatic reactions showed that the enantioselectivities of the hydrolysis of not only the monoesters 3 but also the diesters 4 are almost perfect under the stated reaction conditions. Interestingly, the ee of (S)-2 derived from 4a (Entry 1), which has the lowest number n, was relatively low (83%), and almost the same result was obtained in the case of 4f (80%), which has the highest number n (Entry 6). In these cases, longer reaction times (48 h) did not improve the conversions and the ees of 4, although the reason was not clear yet. These results indicated that the enzyme prefers the substrates bearing a moderate carbon number between two carboxylates in the first enzymatic hydrolysis, and the higher reaction rates of 4b (n = 3, Entry 2) and 4c (n = 4, Entry 3) caused the higher ees of (S)-2 derived from 4b and 4c. Finally, we determined that glutarate and adipate were the most suitable substrates (4b and 4c, respectively) for this enzymatic reaction.
We next studied the time-course of the reaction for 4c using a smaller amount of enzyme (40 mg for 0.4 mmol of substrate), and the results are shown in Table 2.
![]()
Table 1. Enantioselective hydrolysis of dicarboxylic diesters mix-4a-f with Novozym 435a.
aThe reaction was performed using the substrate (4.00 mmol) with Novozym 435 (80 mg) in 0.1 M phosphate buffer (pH 6.5, 40 mL) for 24 h at 30˚C. bDetermined by GC analysis of (S)-2 after chemical hydrolysis. cDetermined by GC analysis.
![]()
Table 2. Enzymatic enantioselective hydrolysis of dicarboxylic diester mix-4ca.
aThe reaction was performed using the substrate (4.00 mmol) with Novozym 435 (40 mg) in 0.1 M phosphate buffer (pH 6.5, 40 mL) at 30˚C. bDetermined by GC analysis of (S)-2 after chemical hydrolysis. cDetermined by GC analysis.
Beyond our expectation, the reaction for only 1 h smoothly proceeded to afford the optically pure (R)-2 in 59% yield (Entry 1). In the case of the reaction for 12 h, the yield of (R)-2 reached 90%, and the ee of (S)-2 from the monoester 3c was >99% (Entry 2). According to the slightly lower ee (97%) of (S)-2 from the diester 4c, it was proposed that the enantioselective hydrolysis of 4c should be slower than that of 3c. Finally, the reaction for 24 h gave the complete resolution (Entry 3). The yields of the products (R)-2, (S)-3c, and (S, S)-4c were 96%, 50%, and 25%, respectively, and the ees of all the compounds 2 were over 99%. This reaction was also useful in a preparative-scale operation (1.43 g of 3c) using an Erlenmeyer flask for 24 h at 30˚C. We obtained (R)-2 (>99% ee) in 100% and (S)-2 (>99% ee) in 88% total isolated yields from (±)-4c.
3.4. Enzymatic Hydrolysis of Various Dicarboxylic Diesters
In order to apply the concept of this reaction to the kinetic resolution of other secondary alcohols, we next examined the enzymatic hydrolysis of several glutarates and adipates (18 - 23; n = 3 and 4, respectively), and these results are shown in Table 3. In the cases
![]()
Table 3. Enzymatic enantioselective hydrolysis of dicarboxylic diesters mix-18-23a.
aUnless otherwise noted, the reaction was performed using the substrate (4.00 mmol) with Novozym 435 (40 mg) in 0.1 M phosphate buffer (pH 6.5, 40 mL) at 30˚C. bDetermined by GC or HPLC analysis of the corresponding (S)-alcoholafter chemical hydrolysis. cDetermined by GC or HPLC analysis. dThe reaction was performed in the mixed solvent of 0.1 M phosphate buffer (pH 6.5) containing 10% iPr2O. eThe reaction was performed in the mixed solvent of 0.1 M phosphate buffer (pH 6.5) containing 25% iPr2O.
of entries 1-8, the reactions were performed for a longer reaction time (48 h) because the reactivities were lower than that in the case of the substrate 4.
The hydrolyses of both the glutarate mix-18b and the adipate mix-18c bearing an ethyl group as the R2 substituent (R1 = Ph, R2 = Et) smoothly proceeded with excellent enantioselectivities to afford the optically pure alcohol (R)-6. However, in the case of Entry 1, the hydrolysis of the monoester 12b was slower than that of the diester 18b, and then the ee of monoester (S)-12b was low. This tendency was quite different from that in the case of 4 previously mentioned. In the cases of mix-19b and 19c bearing a phenylmethyl group (R1 = PhCH2, R2 = Me), the enzyme completely discriminated the enantiomers to afford the optically pure products in a manner similar to 4 (entries 3 and 4). Although the diesters mix-20b and 20c bearing a 2-naphthyl group (R1 = 2-naphthyl, R2 = Me) were slowly hydrolyzed (entries 5 and 6), the enantioselectivities were excellent and the optically pure (S)-14 and (R)-8 were obtained. On the other hand, both the reactivities and enantioselectivities of the diesters (mix-21b and 21c; R1 = 1-naphthyl, R2 = Me) of 1-(1-naphthyl)ethanol (9) were extremely low (entries 7 and 8). These results indicated that the 1-naphthyl group would be too bulky for the interaction between the substrate and the enzyme. Interestingly, the enzymatic reactions of the diesters mix-22 b, 22 c and mix-23b, 23c, which contain a phenylethyl (R1 = PhCH2CH2, R2 = Me; entries 9 and 10) and benzyloxyethyl group (R1 = BnOCH2CH2, R2=Me; entries 11 and 12), respectively, smoothly proceeded for only 24 h with sufficient enantioselectivities to give the corresponding optically active compounds. It is noteworthy that the ees of the monoesters (S)-17b and 17c were higher than those of the alcohol (S)-11 derived the remaining diesters 23b and 23c, and the resulting alcohols (R)-11 were not of the optically pure form. In our previous report, the kinetic resolution of the monoester (±)-17b with Novozym 435 was completely accomplished to afford the almost optically pure alcohol 11 (E value = 920) under the same reaction conditions. These results indicated that the enantioselectivities of the first enzymatic hydrolyses of mix-23b and 23c should be lower than those of the second reactions of (±)-17b and 17c.
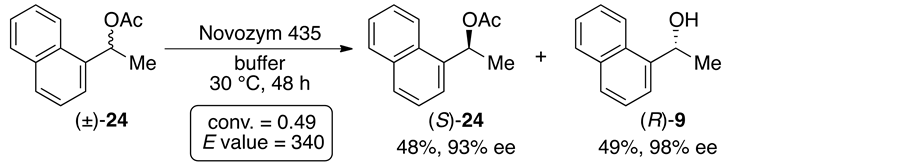
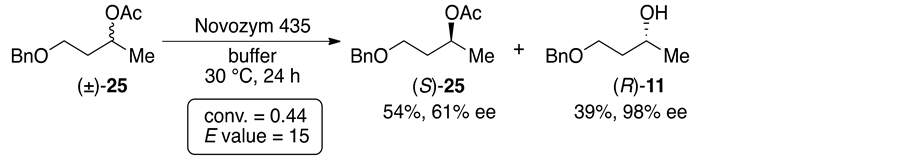
For comparison, we also examined the enzymatic hydrolysis of the usual acetates derived from the corresponding alcohols 2 and 6 - 11 under the same reaction conditions as mentioned above. Among the reactions of all the acetates, the reactivitiesand/ore- nantioselectivities in the cases of the acetates (±)-24 and 25 bearing a 1-naphthyl group and a benzyloxyethyl group, respectively, were quite different from those of the dicarboxylic acid diseters 21 and 23 containing the same substituents, while other acetates were enantioselectively hydrolyzed in a manner similar to the corresponding dicarboxylic acid diseters. Surprisingly, in the case of 24 (R1 = 1-naphthyl, R2 = Me), the reaction was smoothly accomplished to afford optically active compounds (conv. = 0.49, E value = 340; Scheme 6(a)) [15] . On the other hand, the enantioselectivity in the case of 25 (R1 = BnOCH2CH2, R2 = Me) was low, although the hydrolysis smoothly proceeded (conv. = 0.44, E value = 15; Scheme 6(b)). These results indicated that the structure of
(a)
(b)
Scheme 6. Enzymatic hydrolysis of the acetates (±)-24 and 25 with Novozym 435.
the acyl moieties apparently affects the interaction between the substrates and the active site of the enzyme, and the use of the dicarboxylic acid diseters as the substrates could bring the latent specificity of molecular recognition in enzymatic hydrolysis.
4. Conclusion
In this study, we succeeded in the enzyme-mediated enantioselective hydrolysis of aliphatic dicarboxylic acid diesters, and obtained several enantiomers of 2, 6, 7, 8, 9, 10 and 11. We also disclosed that the reactivity and enantioselectivity could be controlled using a suitable acyl group of the substrates, and the glutarate and adipate were suitable as the acyl moiety of the substrates. Furthermore, we found that the substrate specificity of the enzyme differed from those in the case of the corresponding acetates. We anticipate that the use of glutarate and adipate for the enzymatic hydrolysis could be an alternative choice as the simple acetates.
Acknowledgements
We thank Collaborative Research Center (Meisei University) for the analyses of organic compounds, and Mr. Masamune Minegishi (Meisei University) for helpful discussions.