Theoretical Study of the Interaction between Chitosan Constituents (Glucosamine and Acetylglucosamine Dimers) and Na﹢ Ions ()

Subject Areas: Chemical Engineering & Technology
1. Introduction
The functioning of any biological molecule depends fundamentally on the shape and flexibility, and further on interactions with neighboring environment, interactions provide feedback on their preferred structures and preferred bio-active conformer as well [1] [2] . For that reason several experimental and theoretical studies about interactions of different molecules have been done where different methods have been employed like nucleation, growth in liquid or gas phase to mention but a few [1] . Molecular structure and interactions lie in the area of molecular physics; it generally involves experimental methods like electronic and vibrational spectroscopy, mass spectrometry, and other techniques. The theoretical DFT calculations of electronic structure and force field can lead to structural assignment and more importantly, identify the interactions within the isolated molecules and between molecules and environment [3] [4] . An interesting feature and advantage of the DFT method is that it specifies the simplest structure which can demonstrate intricacies and challenges of larger and complex systems [5] . DFT provides theoretical scrutiny of the spectra to weigh against experiment, the structural view which they reflect, and the non-covalent interactions that control them [3] [6] [7] .
Chitosan is a harmless and eco-friendly polysaccharide that has in recent times emerged as a promising contender for diverse applications [8] [9] . Different chitosan constituents conformers exist and they differ in the fractional content of acetylated units and the degree of polymerization [8] [10] [11] . Chitosan, a linear co-polymer of glucosamine and N-acetylglucosamine in a β1-4 linkage obtained by N-acetylation of chitin [11] [12] , is a biodegradable polysaccharide with a wide range of biomedical applications such as sutures, wound dressing, bone substitutes, tissue engineering, and gene and drug delivery systems [3] [6] [10] [13] . It is a structural material that is nowadays commonly used in chemistry, medicine, and in various technologies [2] [6] . It is biocompatible, biodegradable, nontoxic, nonflammable, nonallergenic, antimicrobial, and inexpensive [2] [13] - [18] . The chitosan constituents, glucosamine and acetylglucosamine, play an important role in multifarious applications in different fields like pharmaceutics, cosmetics, biomedicine and agriculture to mention but a few.
Previous computation exertions had been done about interaction of chitosan constituents with heavy metals, alkali and halide ions [3] [19] . The present study is the continuation of our previous work [20] where two different conformers of glucosamine monomer named AX and AY and acetylglucosamine B as well as their interraction with sodium ion were considered. Here we attempt to move a little further and investigate the interaction between dimers of glucosamine and acetylglucosamine (AXAX, AYAY, and AYB) with sodium ion. The aim of the this work is to determine the geometrical structure, vibrational spectra, and thermodynamic properties of the dimer molecules, AXAX, AYAY, and AYB, as well as their adducts with sodium ion, AXAXNa+, AYAYNa+, and AYBNa+, and the enthalpies of the association reactions between Na+ and the dimers.
2. Computational Details
The DFT/B3LYP method under the 6-31G(d) basis set was applied for the calculations of the equilibrium geometrical parameters and vibrational spectra of dimers AXAX, AYAY, AYB and their complexes with Na+. Embarking on this work an ensemble of low energy conformers of interest were generated using HyperChem program package [21] . Then the designed configurations were used in the optimization procedure with the Firefly software [22] partially based on the GAMESS (US) source code [23] . Vibrational spectra calculated proved the structures to be at equilibrium by the absence of imaginary frequencies. The energies of the reactions were computed with several basis sets from 6-31G(d) up to 6-311++G(d, p). For the visualization of the optimized structures, and specification of geometrical parameters and vibrational modes, the wxMacMolplot [24] and Chemcraft [25] softwares have been applied.
3. Results and Discussion
3.1. Geometrical Structure of Dimers and Adducts
The equilibrium structures of the AXAX, AYAY, AYB and adducts AXAXNa+, AYAYNa+, and AYBNa+ are shown in Figure 1, Figure 2. The selected geometrical parameters and atomic charges are given in Table 1, Table 2. The dimers and adducts comprise two rings, each of the ring made up of five carbon and one oxygen atom inserted between. These rings are fixed jointly by the glycosidic bond, the bond which ensures the stability and stiffness of this molecule [7] [10] . The glycosidic bond bends the rings in opposite direction extricating rings from each other and thus reduces repulsive forces flanked by the two.
AXAX and AXAXNa+. In the AXAX dimer, glycosidic bond forms an angle C2-O12-C15 = 119˚ in Figure 1(a), and Table 1, whose respective length C2-O12 and C15-O12 are 1.438 Å and 1.400 Å correspondingly. In the AXAXNa+ complex the geometrical parameters are not changed drastically, the angle C2-O12-C15 is 118˚ the bond length C2-O12 is 1.426 Å and C15-O12 is 1.412 Å. That is the attachment of Na+ to the dimer does not alter significantly the shape of the glycosidic bond and this is because Na+ attaches at the distance far away from the bond. In the AXAX molecule hydroxyl group and amine group are in opposite sides to each other to reduce mutual repulsion of like charges. Amine groups are located in rather big distance between, 7.175 Å, thus the stable conformer of AXAX is formed. In the rings of AXAX the internal C-O-C groups exist, in the first ring
![]()
![]() (a) (b)
(a) (b)
Figure 2. Equilibrium geometrical structure of the species: AYB (a) and AYBNa+ (b).
![]()
Table 1. Selected geometrical parameters and atomic charges of AXAX and AXAXNa+.
![]()
Table 2. Selected geometrical parameters and atomic charges of AYB and AYBNa+.
C4-O7-C5 angle is 114˚, its respective bond lengths are C4-O7 = 1.419 Å and C5-O7 = 1.433 Å, in the other ring the C15-O18-C16 angle is the same and the bond lengths are 1.423 Å for C15-O18 and 1.439 Å for C16-O18. That is the internal C-O-C fragments are alike in both rings of AXAX implying to have the same chemical environment before Na+ attachment.
In AXAXNa+ adduct, the position of the Na+ connection is ascribed to the negative charges of the N11, O1 and C4 (Table 1) and appropriate internuclear separations. The attachment of Na+ creates two new bonds Na+-O1 and Na+-N11. Compared to AXAX, in AXAXNa+ adduct the attached Na+ results in some modification to the original structure, the C4-O7-C5 = 117˚ in the first ring whose respective bond lengths are C4-O7 = 1.382 Å and C5-O7 = 1.448Å, and the next ring C15-O18-C16 = 116˚ whose respective bond lengths are 1.417 Å for C15-O18 and 1.445 Å for C16-O18. Furthermore in the adduct AXAXNa+, the internal hydrogen bond O8 - H29…O18 (1.853 Å) is formed which increases the adduct stability and stiffness.
AYAY and AYAYNa+. In the dimer AYAY the amine groups are separated by distance of 4.979 Å that is significantly less as compared to AXAX (7.175 Å). This may lead to decrease in the stability of the dimer AYAY because of the repulsive force experienced between amine groups. Also this dimer has two hydrogen bonds O10-H32…O8 and O20-H43…O22 whose respective lengths are 1.865 and 1.820 Å. The formation of these internal hydrogen bonds favors the stability of the dimer. In the glycosidic bond of AYAY, the angle C5-O9-C13 is equal to 117˚ and the bond lengths C5-O9 and C13-O9 are 1.438 Å and 1.398 Å correspondingly (Table 1) while in the AYAYNa+ complex, the similar angle C5-O9-C13 is 115˚ and the bond lengths are C5-O9 = 1.453 Å and C13-O9 = 1.406 Å. Thus akin to AXAXNa+ the attachment of Na+ to AYAY does not bring essential change in the parameters of the glycosidic bond. At the same time contrary to AXAXNa+, the position of Na+ connection to AYAY is close to the glycosidic bond; it attaches to two oxygen atoms (one of the glycosidic bond and another of the ring) and to the nitrogen atom connected with the second ring.
In AYAYNa+ adduct, the position of the Na+ attachment is credited to the negative charge −0.39 of the N12 in the dimer AYAY and the appropriate internuclear separations and steric factors as well. Three new bonds are formed: Na+-O9, Na+-O18 and Na+-N12 with internuclear separations 2.335 Å, 2.253 Å, and 2.347 Å, respectively. Two hydrogen bonds O10-H32…O8 and O20-H43…O22 remain in adduct AYAYNa+, and their respective lengths do not change remarkably as compared to AYAY.
AYB and AYBNa+. The equilibrium structures of the AYB complex and the AYBNa+ adduct are shown in Figure 2; and the selected geometrical parameters and atomic charges are given in Table 2. In the glycosidic bond of AYB connecting two rings the angle C2-O12-C15 is equal to 118˚, and bond lengths are C2-O12 = 1.438 Å and O12-C15 = 1.408 Å. In the AYBNa+, the C2-O12-C18 angle is 119˚ and the respective bond lengths C2-O12 and O12-C15 are 1.457 Å and 1.378 Å, thus slight change in the C-O-C parameters are noticed when Na+ is attached. In AYBNa+, three bonds are formed between Na+ and negatively charged oxygen atoms O18, O21, O22. In AYBNa+ adduct the amino groups are located far apart at the distance of 7.244 Å between two nitrogen atoms to ebb electron-electron repulsion. Whereas in AYB molecule there is no internal hydrogen bond, and in its adduct the hydrogen bond N23-H47…O10 is formed; this connotes that attachment of Na+ stabilizes the species.
3.2. Vibration Spectra of Dimers and Adducts
In the calculated vibrational spectra there were no imaginary frequencies revealing the equilibrium geometrical structures of the species. The theoretical spectra of the dimer molecules and adducts with Na+ ion are presented in Figures 3-5. The general feature of all these spectra is an existence of three regions of vibrational modes, which are approximately (i) below 1700 cm−1, (ii) about 3000 cm−1, and (iii) above 3300 cm−1. The assignment of the most intensive peaks is considered below.
AXAX and AXAXNa+. The vibrational spectra of AXAX and AXAXNa+ adducts are shown in Figure 3. As is seen, majority of bands are located at a low frequency region 100 - 1690 cm−1 which correspond to O-H rocking, NH2 scissoring, and ring bending vibrations. For the dimer AXAX the strongest peak seen at 1067 cm−1 is ascribed to bending of the first ring, and the subsequent peak at 1352 cm−1 is assigned to the bending of the second ring. An intricate vibration mode at 717 cm−1 relates to the bending of different fragments of the molecule. The middle region ranges from 2900 - 3100 cm−1 in both AXAX and AXAXNa+ adduct; the most intensive peak relates to C-H stretching at about 3034 cm−1. The higher frequency region ranges between 3400 - 3750 cm−1 for both AXAX and the AXAXNa+ adduct where very intense O-H stretching modes are seen at about 3500 cm−1 and 3474 cm−1 for AXAX and AXAXNa+, respectively. Also the peaks at 3581 cm−1 (AXAX) and 3444 cm−1 (AXAXNa+) observed are assigned to the combination of O-H and N-H stretching modes.
AYAY and AYAYNa+. The vibrational spectra of AYAY and AYAYNa+ are shown in Figure 4. Majority of the vibrational bands manifests at the first frequency region. The most intensive vibrational peak is seen at 1089 cm−1 associated to bending of the ring, other peaks are seen also at 186 cm−1 relating to O-H rocking and ring bending, at 800 cm−1 NH2 wagging, O-H rocking and a vibration band at ~1670 cm−1 assigned to the NH2 scissoring. The similar bands are seen in AYAYNa+ adduct. In the middle region at about 3000 cm−1 C-H stretching vibrational modes are seen for both AYAY and AYAYNa+ adduct; and in the higher frequency region O-H and NH2 stretching vibrations may be observed between 3400 - 3750 cm−1 in both cases.
AYB and AYBNa+. In the IR spectra of AYB and AYBNa+ the most intensive bands are seen in low and high frequency regions (Figure 5). In the spectrum of AYB molecule, the peak at 1741 cm−1 is assigned to a combination of C=C bond stretching and N-H rocking, the next band at 1035 cm−1 corresponds to the combination of C-O stretching and ring bending. Other peaks are ascribed to O-H rocking, N-H scissoring, and knotty vibration mode of the ring. In the middle region the C-H stretching vibration is observed at 3019 cm−1 for AYB and at 3061 cm−1 for the adduct. Lastly in the third region, N-H, and O-H stretching vibration modes of the AYB are
![]() (a)
(a)![]() (b)
(b)
Figure 3. Theoretical IR spectra of AXAX (a) and AXAXNa+ (b).
![]() (a)
(a)![]() (b)
(b)
Figure 4. Theoretical IR spectra of AYAY (a) and AYAYNa+ (b).
![]() (a)
(a)![]() (b)
(b)
Figure 5. Theoretical IR spectra of AYB (a), and AYBNa+ (b).
seen between 3391 - 3668 cm−1 while for AYBNa+ adduct the frequency 3359 cm−1 is assigned to the stretching vibration of NH…O (hydrogen bond).
Concluding this section on the spectra of the dimer molecules and the adducts with Na+ ion, some similarities may be noted which are apparently due to the same functional groups of atoms, NH2, CH, OH, COC, and other fragments, and the structural resemblance of the species as well. In the AANa+, and ABNa+ adducts, the Na+ ion only participates in low frequency vibrations, less than 395 cm-1 of low IR intensity. Worth to mention here that the findings of this study are in accordance with the theoretical spectra of the glucosamine molecules A, AA, and AAA [7] , and ANa+ and BNa+ adducts [20] as well with the experimental IR spectra of chitosan [26] [27] .
3.3. Thermodynamics of Association Reactions
The reactions of the sodium ion attachment to the dimer molecules were considered:
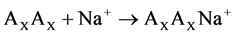 (1)
(1)
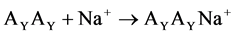 (2)
(2)
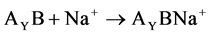 (3)
(3)
The energies of the reactions DrE were calculated as the difference between the total energies of the product and reactants:
![]() (4)
(4)
The enthalpies of the reactions ∆rH°(0) were obtained using ∆rE and the zero-point vibration energy corrections ∆ZPVE:
![]() (5)
(5)
![]() (6)
(6)
where h is the Plank’s constant, c is the speed of light in the free space, ![]() , and
, and ![]() are the sums of the vibration frequencies of the product and reactants, respectively.
are the sums of the vibration frequencies of the product and reactants, respectively.
The enthalpies of the reactions (1)-(3) have been calculated under different basis sets, 6-31G(d), 6-31G(d, p), 6-311G(d), 6-311G(d, p), 6-31++G(d, p) and 6-311++G(d, p); the results are presented in Figure 5. For all three reactions, the similarity of the plots is observed. The general trend observed reveals the increase of the enthalpies of the reactions with the basis set extension. The difference between the lowest and highest values of ∆rH°(0) for each reaction is about 50 kJ∙mol−1, thereafter the uncertainties for the enthalpies of the reactions may be estimated as half of this difference, that is ~25 kJ∙mol−1. The values energies ∆rE, ∆ZPVE, and enthalpies ∆rH°(0) of the reactions obtained with the most extended basis set 6-311++G(d, p) are given in Table 3. The results indicate the association processes are highly exothermic, that implies a strong interaction between the cation and dimer molecules and high energetic stability of the corresponding adducts formed. The data obtained here are close to the enthalpies of attachment of sodium ion to monomer molecules, −196 kJ∙mol−1 (AX + Na+ = AXNa+), −227 kJ∙mol−1 (AY + Na+ = AYNa+), and −229 kJ∙mol−1 (B + Na+ = BNa+) [20] . Apparently likeness of these results relates to the similarity of the interacting moieties of the monomer or dimer molecules with Na+ ion.
The Gibbs free energies ∆rG°(T) of reactions (1)-(3) were calculated using the following equation:
![]() (7)
(7)
where ∆rH°(T) is the enthalpy of the reaction at temperature T, ∆rS°(T) is the change in entropy of the reaction. The thermodynamic functions of the AXAX, AYAY, AYB, AXAXNa+, AYAYNa+ and AYBNa+, have been computed using the OpenThermo software [28] , those for Na+ gaseous ions are taken from [29] . The geometrical parameters and vibrational frequencies needed for the thermodynamic functions calculations have been obtained under 6-31G(d) basis set. The thermodynamic functions of the species are given in Table 4 for the temperature
![]()
Table 3. The reaction equations, energies ∆rE, zero point vibrational energy corrections ∆ZPVE and enthalpies ∆rH°(0) of the reactions; in kJ∙mol−1.
![]()
Table 4. Thermodynamic functions of AXAX, AYAY, AYB dimers and AXAXNa+, AYAYNa+ and AYBNa+ adducts.
aΦ°(T) = −[H°(T) − H°(0) − TS°(T)]/T is the reduced Gibbs free energy.
range between 298 K and 1000 K. To find the enthalpies ∆rH°(T), the enthalpy increments H°(T) − H°(0) were used:
![]() (8)
(8)
where the values of ∆rH°(0) were obtained using Equation (5) and the values of ∆rE calculated with 6-311++G(d, p) basis set.
The plot of the Gibbs free energies of reactions (1), (2) and (3) are presented in Figure 6. The values of ∆rG°(T) are negative in a broad temperature range, this indicates that the attachment processes are spontaneous; ∆rG°(T) become positive at temperatures higher than 1700 K for the reaction (1), and 2000 K for (2) and (3). For the reverse process it is evident that their corresponding adducts are stable with respect of the detachment of Na+ ion in a broad temperature range (Figure 7).
![]()
Figure 6. The calculated enthalpies of the reactions (1)-(3) vs. basis set.
![]()
Figure 7. Gibbs free energy vs. temperature for the reactions AXAX + Na+ = AXAXNa+, AYAY + Na+ = AYAYNa+ and AYB + Na+ = AYBNa+, 6-311++G(d, p) basis set.
4. Conclusions
The attachment of Na+ ion to the dimers AXAX, AYAY, AYB to form adducts has been studied using the DFT/B3LYP method. The geometrical structures, vibrational spectra of the dimer molecules and their corresponding adducts due to Na+ attachment have been determined. Position of Na+ attachment is dictated by negative atomic charges occurrence, appropriate internuclear separations and steric factors. In the adducts AXAXNa+ and AYAYNa+, the sodium ion is linked to nitrogen and oxygen atoms; while in the AYB molecule nitrogen atoms are screened by the acetyl groups, and therefore Na+ sticks to three oxygen atoms instead.
The scrutiny of the calculated IR spectra revealed the general feature for all species, which is an existence of three regions composed of the similar vibrational bands which have been assigned to the certain functional groups of atoms, NH2, CH, OH, COC, and other fragments. The structural resemblance of the species also has been brought to the similarity of the spectra. Thermodynamic characteristics of attachment have been determined. The exothermicity and spontaneity character of adducts formation have been ascertained.
Acknowledgements
The authors are grateful to The Nelson Mandela African Institution of Science and Technology, and The British Gas (BG group) for the sponsorship. In a special way we acknowledge a valuable assistance and service by the School of Computational and Communication Science and Engineering.
NOTES
![]()
*Corresponding author.