1. Introduction
Soil gas is defined as the atmosphere present in pore spaces of soil matrices [1] . These pores can undergo contamination phenomena, due to exposure to volatile pollutants. The latter can derive from different sources: contaminated soils and ground waters. Chemical-physical, geological and hydrogeological characteristics of each soil make its contamination highly compound- and site-specific. Therefore, one of the most recent aims of the analytical and environmental chemistry consists in hydrocarbons determination in soil gas matrices and their speciation.
Hydrocarbons represent a class of numerous and heterogeneous organic compounds ranging from linear, cyclic and branched saturated and unsaturated alkanes to aromatic and polycyclic ones. Each of these species is characterized by a peculiar value of toxicity and, as a consequence, their effect on the human health and environment is strongly different. More in detail, the speciation consists in the determination and quantification of hydrocarbons fractions in soil gas matrices.
In order to evaluate hydrocarbons carcinogenic effects, some specific target molecules have been identified: polycyclic aromatic hydrocarbons (PAHs), benzene, etc.
On the contrary, for toxic non-carcinogenic effects, specific hydrocarbons fractions are taken into account (e.g., in Italy for different environmental matrices C5-C8 aliphatic, C9-C12 aliphatic, C9-C10 aromatic, C11- C12 aromatic, C13-C18 aliphatic, C13-C22 aromatic and C19-C36 aliphatic have been identified) [2] .
If hydrocarbons speciation in contaminated soil and water matrices can be easily analyzed by the use of official analytical methods, the discussion about the more appropriate method for the analysis of soil gases collected on different supports is still open.
Among the official methods [3] -[5] , one is based on hydrocarbon aromatic fraction quantification by extraction of characteristic ions and hydrocarbon aliphatic fraction quantification by total ion integration and subsequent subtraction of internal standard and target compounds peaks. This method is valid up to C12 aliphatic compounds and up to C10 aromatic compounds and is related to the use of canisters in the absence of any extraction solvent [5] .
Another official method is useful for soil and water matrices [3] . It is based on the use of traditional detectors (FID and PID) that discriminate the different hydrocarbons fractions only on the base of the retention time and do not provide any information on the chemical species present in the samples.
Our needs, however, require not only the determination of C11-C12 aromatic hydrocarbons and C13-C18 aliphatic fractions (higher than C12), but also the use of CS2 as an extraction solvent to elute hydrocarbons fraction from soil gas matrices sampled on solid sorbent tubes (coconut shell charcoal).
Therefore, in order to develop a unique, even empirical method for this purpose, in this work we demonstrate that gas-chromatography coupled with mass spectrometry is an effective technique for the speciation of hydrocarbons collected on chemical desorption solid sorbent tubes.
2. Experimental
2.1. Materials and Instruments
A standard solution containing all analytes (1000 µg/mL for each analyte: n-pentane, 2-methylpentane, 2,2,4- trimethylpentane, n-nonane, n-butylcyclohexane, n-decane, 1,2,4-trimethylbenzene, naphthalene, butylbenzene, pentylbenzene, biphenyl, n-tetradecane, n-hexadecane, n-octadecane; Solution A) and an internal standard solution (ISTD, 2,5-dibrometoluene, 5000 µg/mL) were purchased by CPAChem and used as received. They were bought as solutions in CS2. CS2 (99.9% purity low benzene) and solid sorbent tubes (coconut shell charcoal) for chemical desorption process were purchased by Sigma Aldrich. Real samples (SAMPLE 1-8), contaminated with aliphatic hydrocarbons, were sampled on solid sorbent tubes reported above.
All samples were analyzed by a GC/MS technique using an Agilent 7890A GC equipped with a 5975C inert MSD (mass spectrometer detector) with triple axis detector. The Agilent 7890A GC was interfaced with an Agilent 7683B Series Injector―Automatic Liquid Sampler. A Thermo TR5-MS (60 m × 0.25 mm × 1 µm) column was employed.
2.2. Certified Reference Materials (CRM) Preparation for Calibration Method
Calibration standards were prepared by dilution of Solution A with a diluted internal standard solution (d-ISTD, with a concentration of 10 μg/mL in CS2), as described in Table 1.
At each STD in the Table 1 corresponds a calibration level (CAL LEV).
All the STD solutions reported in Table 1 were injected in a GC/MS instrument interfaced with an automatic liquid sampler (Section 2.5).
2.3. Fortified Materials Preparation for Method Validation (Precision and Recovery)
The availability of certified reference materials (CRM) is limited [6] . Moreover, because CRM are not commercially available for this kind of analyses (sample of solid sorbent tube coconut shell charcoal), fortified materials were prepared in-house. For this purpose, starting from Solution A, STD 8 and STD 5 were prepared as reported in Table 1 but using pure CS2 instead of diluted internal standard solution. Therefore, 100 μL of Solution A, STD 8 and STD 5 were spiked on 200 mg of coconut shell charcoal (solid sorbent tube).
This procedure was repeated ten times for each solution in order to obtain a series of ten samples for each concentration.
2.4. Fortified Materials Analysis from Chemical Desorption of Sorbent Tubes
All materials reported in the Section 2.3 were extracted with 2 mL of d-ISTD (1 mL of d-ISTD for each 100 mg of carbon) [7] .
2.5. GC/MS Analytical Procedure
All samples were processed using the following GC/MS conditions.
Oven Program: 40˚C for 8 min; then 4˚C/min to 100˚C for 0 min; then 15˚C/min to 320˚C for 9 min; then 40˚C/min to 330˚C for 1 min. Injection Volume: 1 µL. SS Inlet: Mode splitless. Heater: 250˚C. Column: TR5- MS 60 m × 250 µm × 1 µm. Flow: 1.2 mL/min. MSD Parameters: MS Source 230˚C, MS Quadrupole 150˚C. SIM Parameter: characteristic ion Target and Qualifier for each VOC compounds. SCAN Parameter: Low Mass: 35.0 amu; High Mass: 350 amu. SIM and SCAN modes were simultaneously acquired.
3. Results and Discussion
Starting from the data reported in the official methods [3] [5] and on the base of our experience and technical requirements, an accurate mass spectra investigation of numerous chemical species representative of each hydrocarbons fraction (C5-C8 aliphatic, C9-C12 aliphatic, C9-C10 aromatic, C11-C12 aromatic, C13-C18 aliphatic) was carried out.
Table 2 reports standard markers and corresponding ions (m/z) used to quantify each hydrocarbons fraction, as well as the hydrocarbons range in terms of retention time.
All chromatograms were contemporary acquired by SIM―(selected ion monitoring) and SCAN-mode. SIM- mode analysis allows the quali-quantitative determination of target compounds, such as BTEX (benzene, toluene,
![]()
Table 1. Calibration standards (STD) preparation and hydrocarbons concentration in each fraction.
![]()
Table 2. Target ions (m/z), standard markers and retention time window for each hydrocarbons fraction. *From Ref. [5] .
ethylbenzene, xylenes), MTBE (methyl-ter-butyl-ether), halogenated VOC (volatile organic compounds), etc. SCAN-mode analysis permits the identification and determination of hydrocarbons fractions by characteristic ions extraction (SCAN-ion quantification) within the typical retention time of each fraction.
SCAN-ion approach allows achieving many benefits. Among them, the most important one consists in a correct and easy integration of more critical fractions: C5-C8 and C13-C18 aliphatic.
In fact, C5-C8 aliphatic fraction could be affect by the presence of solvent front (CS2), whereas column bleed, strictly related to the oven temperature increase, could have a negative effect on the C13-C18 aliphatic fraction determination, in particular at low concentration, causing an analytical sensitivity drop.
SCAN-ion approach, based on the characteristic ions extraction, improves analytical performances, as described in Figure 1.
3.1. Initial Calibration
The relationship between detector response of each analyte and collective range concentration was calculated by the RRFs approach (Equation (1)):
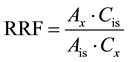 (1)
(1)
where RRF = relative response factor, Ax = sum of ions areas of each fraction to be measured, Cis = concentration of internal standard spiking mixture, µg/mL, Ais= area of the primary ion for the internal standard, Cx = concentration of hydrocarbons fraction in the calibration standard, µg/mL.
The calculated %RDS (Equation (2)) the RRF for each hydrocarbons fraction in the calibration method resulted to be less than 30%, as required by the technical acceptance criteria for initial calibration [5] .
![]()
Figure 1. (A) (B) SCAN-mode acquired chromatograms and hydrocarbon fractions quantification using total ion integration; (C) (D) SCAN-mode acquired chromatograms and hydrocarbon fractions quantification using SCAN-ion approach by characteristic ions extraction (m/z 57, 71, 41).
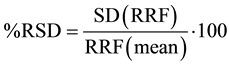 (2)
(2)
where SD (RRF) = standard deviation of initial response factors for each fraction, RRF(mean) = mean of initial relative response factors for each fraction.
However, when these technical criteria were not satisfied, linear and nonlinear regressions were used, as for the C5-C8 aliphatic fraction (linear regression R2 ≥ 0.98).
Moreover, following the recommendations reported in [8] , other technical acceptance criteria, were satisfied, as:
・ The RRT (Relative Retention Time) for each hydrocarbons fraction at each calibration level was within 0.06 RRT units of the mean RRT for the fraction;
・ The Retention Time (RT) shift of ISTD at each calibration level was within 20 s of the mean RT over the initial calibration range for ISTD;
・ The area response of the internal standard at each calibration level was within 40% of the mean area response over initial calibration range for ISTD.
3.2. Precision, Recovery and Uncertainty
As described in Section 2.3, ten fortified materials at three different concentrations were prepared, in order to evaluate quality data of the method.
Tables 3-5 summarize mean of precision data, CV% and recovery% obtained for all fractions at three concentrations (µg/mL) reported in the Table 1.
It is worth to notice that in all data reported in Tables 3-5 CV% and recovery% are in line with the technical acceptance criteria reported in the literature [5] .
All data were analyzed by means of statistical tests: Shapiro-Wilk normality test and Dixon test for identify and reject outliers.
![]()
Table 3. Mean value, CV% and recovery% for each hydrocarbons fraction at concentration corresponding to STD 1 in Table 1.
![]()
Table 4. Mean value, CV% and recovery% for each hydrocarbons fraction at concentration corresponding to STD 4 in Table 1.
![]()
Table 5. Mean value, CV% and recovery% for each hydrocarbons fraction at concentration corresponding to STD 7 in Table 1.
As an example, in Figure 2 the distribution obtained for C13-C18 fraction at concentration values corresponding to STD 1 in Table 1 is reported.
The LOQ of the present method for each hydrocarbons fraction was mathematically estimated multiplying the standard deviation values, obtained from fortified materials at concentration values corresponding to STD 1 (see Table 1), by 5 (Table 6).
Uncertainty was estimated by metrological approach calculating each component to the final uncertainty [9] . For all fractions the uncertainty value was estimated about ±60%.
4. Real Samples Analysis
Eight real samples collected on solid sorbent tubes (SAMPLE 1-8), contaminated with C5-C8 and C9-C12 aliphatic hydrocarbons fraction, were quantified by both the traditional TIC-SCAN method [5] and the present one (SCAN-ion), in order to prove the performances of our innovative analytical approach.
It is worth to notice that the results (Table 7, Table 8 and Figures 3-6) obtained by the two methods are rather comparable and correlated with a mean difference that lower than 30%, which is included in the uncertainty.
![]()
Figure 2. Normal distribution of C13-C18 fraction data.
![]()
Table 6. Standard deviation (SD) and limit of quantification (LOQ) for all hydrocarbons fractions.
![]()
Table 7. A comparasion between the C5-C8 aliphatic hydrocarbons fraction concentration of real samples quantified by TIC-scan and SCAN-ion approaches.
![]()
Table 8. A comparasion between the C9-C12 aliphatic hydrocarbons fraction concentration of real samples quantified by TIC-scan and SCAN-ion approaches.
![]()
Figure 3. Trend of SCAN-ion vs TIC-scan for C5-C8 aliphatic hydrocarbons fraction.
![]()
Figure 4. Correlation between data (Table 7) derived by TIC-scan and SCAN-ion approaches.
![]()
Figure 5. Trend of SCAN-ion vs TIC-scan for C9-C12 aliphatic hydrocarbons fraction.
![]()
Figure 6. Correlation between data (Table 8) derived by TIC-scan and SCAN-ion approaches.
5. Conclusions
As an alternative to official methods, in this work an innovative empirical GC/MS method for hydrocarbons speciation in soil gas matrices was proposed.
It is based on the use of a mass spectrometer as the detector and on SCAN-ion acquisition approach that allows separating the different fractions and identifying the chemical species present in all fractions, permits the use of an extraction solvent (CS2) to elute hydrocarbons fraction from solid sorbent tubes (coconut shell charcoal) and extends the determination to hydrocarbons fraction up to C18.
Good performances in terms of precision, recovery and uncertainty were obtained.
The analyses of real samples provided promising results compared to those from traditional approaches. The simplicity and velocity of the method make it a good candidate for wide applications in routine work for analyses of soil gas matrices sampled on solid sorbent tubes.
NOTES
*Corresponding author.