Computational Analysis of Physicochemical, Pharmacokinetic and Toxicological Properties of Deoxyhypusine Synthase Inhibitors with Antimalarial Activity ()
1. Introduction
Malaria is a disease prevalent in tropical climate countries regarded as a major social and economic problems in the world, being considered one of the oldest diseases, affecting a quarter of the world population. Malaria is inserted mainly in third world countries, concentrating on the Americas, Southeast Asia and Central Africa, with the highest death rate in Africa, caused by public health problem in the region [1] . In Brazil there three species responsible for the causes of malaria in humans that are: P. vivax, P. falciparum and P. malariae. A fourth species known as P. ovale, is only found in restricted areas of the African continent, and may occasionally be diagnosed in Brazil [1] .
Many studies on the rational design of new antimalarial drugs have been performed using molecular modeling. However, complex molecular systems containing external and internal transition atoms, proteins, polymers, or compounds with a higher molecular weight overestimate the ability of computational chemistry to obtain molecular properties, which can lead to inaccurate results when compared with experimental data [2] [3] .
The appearance of Plasmodium strains resistant to artemisinin has become necessary to search and development of new targets and drugs using computational tools to combat malaria. The description and identification performed by genome sequencing of several species provides the search for new pharmacological targets. In this context, diverse transporter proteins can act as antimalarial targets [4] . Therefore, eukaryotic initiation factor (eIF-5A), whose activation involves deoxyhypusine synthase that has already been suggested to be antimalarial drugs target [5] .
In this work, a computational investigation of 15 inhibitors of deoxyhypusine synthase target existing in databases such as the BindingDB was carried out [6] (www.bindindb.org/bind/index.jsp) to trace a pattern for the physicochemical properties, pharmacokinetic properties: human intestinal absorption (HIA), cellular permeability (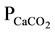 ), cell permeability Maden Darby Canine Kidney (PMDCK), skin permeability (PSkin), plasma protein binding (PPB) and penetration of the blood-brain barrier (CBrain/Blood), and toxicological: mutagenicity and carcinogenicity to the existing inhibitors of this enzyme. Therefore, increase knowledge in this area, and develop new proposals for inhibitors that may be more active and selective, besides to optimizing the investigated properties and decrease side effects.
), cell permeability Maden Darby Canine Kidney (PMDCK), skin permeability (PSkin), plasma protein binding (PPB) and penetration of the blood-brain barrier (CBrain/Blood), and toxicological: mutagenicity and carcinogenicity to the existing inhibitors of this enzyme. Therefore, increase knowledge in this area, and develop new proposals for inhibitors that may be more active and selective, besides to optimizing the investigated properties and decrease side effects.
2. Experimental
2.1. Selection of Molecules and Derivation of the Pharmacophore
Inhibitors of deoxyhypusine synthase target were selected in the database BindingDB, being chosen 15 most active inhibitors, based in Ki value (constant enzyme inhibition), see Figure 1. After this selection process, inhibitors were optimized in Discovery Studio Visualizer 4.0 software (http://accelrys.com) [7] .
The derivation of the pharmacophore was generated with the help of PharmaGist server [8] utilizing 15 inhibitors selected from BindingDB, in order to calculate the pharmacophore group present with the crystallographic inhibitor as a reference (compound 1, Figure 1). The pharmacophore hypothesis is given by the alignment of the most active inhibitors, thereby identifying the regions wherein these are shared [8] . From the identification of each inhibitor (CID) were analyzed in the database BindingDB their respective physicochemical properties related to five rule in accordance with Lipinski parameters [9] .
![]()
Figure 1. Inhibitors of deoxyhypusine synthase target.
2.2. Prediction of Pharmacokinetic and Toxicological Properties of the Inhibitors (ADME/Tox)
The absorption, distribution, metabolism, excretion (ADME) and toxic (Tox) properties were calculated with the aid of online server preADMET [10] . The server calculates parameters such as human intestinal absorption, cellular permeability Caco-2 in vitro, cell permeability Maden Darby Canine Kidney (MDCK), skin permeability, plasma protein binding, penetration of the blood-brain barrier, mutagenicity and carcinogenicity.
2.3. Modeling of New Inhibitors of Deoxyhypusine Synthase Target
Modeling of new inhibitors was performed with the aid of ChemSketch software [11] , from the structural modifications in more active molecule, compound (1) of Figure 1, with the aim of analyze the pharmacokinetic and toxicological properties in an attempt to improve the pharmacological activity with antimalarial activity, being that were proposed three structural modifications, see Figure 2.
In the first modification (compound A) were removed three carbons of the side chain on each side of the molecule from molecule of compound 1 (Figure 1). This modification had the objective of decrease the hydrophobic
![]()
Figure 2. Inhibitors proposed for deoxyhypusine synthase target.
region, providing greater solubility of drug candidate. In the second modification (compound B) was added a hydroxyl (OH−) on carbon atoms 5 and 5’, starting from of most active compound (compound 1). Such modification has the aim to increase solubility, because the hydroxyl group (OH−) is classified as a hydrogen acceptor, enabling the formation of hydrogen bond. In the third amendment (compound C) were removed three carbon atoms from each side of the molecule (carbon 3 and carbon 3’), being replaced by two hydroxyl (OH−) and a double bond in the central nitrogen. The removal of carbon atoms and substitution of hydrogens by hydroxyl had as objective decrease the hydrophobic region creating a higher molecular solubility. Such modifications can alter hydration energy of the molecule.
Hydration energy is released when water molecules are separated from each other and are attracted by molecules or ions of a solute that is dissociating in water, i.e., replacement of the hydrogen atom by the hydroxyl may result in decreased hydration energy resulting in increased solubility of the proposed inhibitors [12] .
The proposed new inhibitors A, B and C were submitted to the ADME/Tox tests and compared with inhibitors selected by database BindingDB.
3. Results and Discussion
3.1. Derivation of the Pharmacophore
The identification of the pharmacophoric groups are defined as essential structural basis for molecular recognition and biological activity. These being important features to propose new drugs [13] .
The derivation of the pharmacophore of deoxyhypusine synthase target was obtained by alignment of 15 molecules, having a score of 22.677, see Figure 3. Compounds aligned share the following characteristics: 3 donor groups and one positive group. Compound 1 (CID3526) is classified as the most active and characteristics are presented inhibitor: 10 hydrophobic groups, 7 donor groups, 1 acceptor group and 2 positive groups, see Figure 4.
3.2. Physicochemical Properties―Lipinski Parameters
A drug to have good oral absorption must satisfy of the parameters following: molecular weight of less than 500 Da, logP (lipophilicity) less than five (5); maximum of five (5) hydrogen donor groups and maximum of ten (10) groups acceptors binding intestinal permeability and comprise the first steps to good oral bioavailability. In analyzing the parameters of the selected compounds was observed that all had values within Lipinski parameters, as shown in Table 1.
![]()
Figure 3. Pharmacophore―alignment of the 15 inhibitors of deoxyhypusine synthase target.
![]()
Figure 4. Pharmacophoric groups of compound 1 (CID3526).
![]()
Table 1. Physicochemical properties for inhibitors of deoxyhypusine synthase target selected in database BindingDB.
3.3. Pharmacokinetic and Toxicological Properties
The results of ADME tests (absorption, distribution, metabolism and excretion) are shown in Table 2. These properties are presented as a determinant for drug development factors, being the biggest target objectives: good absorption, distribution, metabolism and excretion. Mainly, human intestinal absorption properties, because it is determinant for the drug development that purport to be administered orally [13] [14] .
Analyzing the ADME tests of inhibitors shown in Table 2, it was observed that the compounds presented human intestinal absorption values (HIA) in the range of 27.402177 £ HIA% < 89.0000. Being that compounds 2 (HIA = 39.555199%), 7 (HIA = 30.912700%) and 14 (HIA = 27.402172%) showed an absorption low with respect to compound 3 (HIA = 88.386566), having a variation of ±48.831367 (compound 2), ±57.473866 (compound 7) and ±60.984394, respectively. While six compounds (1, 5, 8, 10, 13 and 15) have absorption values in the range of 42.946999 £ HIA% < 60.871844. In relation to compounds (3, 4, 6, 9, 11 and 12) showed the highest human intestinal absorption values, being HIA% ³ 71.643096, highlighted for compound 3 that showed highest degree of HIA with value of 88.386566%, having a variation of ±38.824496% in relation to the compound 1. The absorption processes are related to the permeation of compounds through biological membranes under the influence of physicochemical characteristics [15] .
The cell permeability in vitro Caco-2 is an important test to assess intestinal absorption of drugs. Since, Caco-2 cells derived from human colon adenocarcinoma, having various transport via in the intestinal epithelium [16] . The results of the compounds in Table 2 showed an average permeability of 18.1458. In inhibitor calculated, it was found that the  (nm/s) was moderate, ranging from 7.1205 nm/s to 21.1135 nm/s.
(nm/s) was moderate, ranging from 7.1205 nm/s to 21.1135 nm/s.
The cell permeability in vitro in MDCK system utilizes canine kidney cells and has a shorter growth that the Caco-2 cells. Thus, this system is used as a tool for the rapid analysis of permeability [17] . Analyzing the data in this MDCK system in the inhibitors shown in Table 2, it is verified that the permeability can be classified into low (25 nm/s) and mean (25 to 500 nm/s). Being mean in the compounds (1, 5, 6, 9 and 11) varying between 27.1622 nm/s to 45.4108 nm/s, and low in the other compounds varying between 0.59622 nm/s to 10.4543 nm/s.
The skin permeability is an important factor for the use of the drug transdermal administration via. This parameter is used in the pharmaceutical industry to assess the risk chemical products in case there is accidental contact with skin [18] . All studied and proposed inhibitors showed negative permeability values, not showing importance of use of the drug by this administration via, as shown in Table 2.
![]()
Table 2. Absorption properties of the 15 compounds selected in BindingDB.
[a]percentage of human intestinal absorption; [b]cell permeability (Caco-2 in nm/s); [c]cell permeability Maden Darby Canine Kidney in nm/s; [d]skin permeability.
In Table 3, is shown the human intestinal absorption values for modified compounds. When evaluating the values of HIA was observed that decreased mainly in the compounds B and C which showed a HIA low of 12.505273% and 9.981487%, respectively. However, compound A obtained an absorption classified as mean, presenting equal value to 31.686200% with a range of ±17.875870 in relative to compound 1. The cell permeability in vitro Caco-2 was classified as moderate, but improved relative to compound 1 which had equal value 20.5758 nm/s, while compound A showed a permeability of 21.0928 nm/s. The permeability in MDCK system was considered less than 25 nm/s, mainly in the compound C which showed a permeability of 0.532287 nm/s, demonstrating that the modifications decreased the probability of pharmacokinetic success in the oral administration of possible proposed antimalarial compound.
The drug has two forms in the blood, the plasma protein bond form and non-bond form, although that bond directly depends on their affinity for such bond portion and the free portion. The binding to these proteins can alter the half-life of the drug in the body of the individual [19] [20] .
According to Table 4 of the distribution properties of plasma protein binding (PPB) and penetration of the blood-brain barrier, it is verified that all calculated inhibitors bind poorly to plasma proteins, being this variation of 0.00% to 76.277691%. Compounds 7 and 8 have no force in the bond these proteins by presenting equal value 0.00%, i.e., such inhibitors have a large amount of free drug to interact with their receptors, triggering the form of this pharmacological response. In addition to changing the pharmacological response of molecules the PPB, also modifies the renal excretion, because only unbound drug is available for glomerular filtration, thus increasing its excretion and decreasing the half-life [21] .
The blood-brain barrier (BBB) is an important component of a communication network that connects the central nervous system and peripheral tissues, limiting and regulating the exchange of substances between blood and the central nervous system [22] . Therefore has an importance in the pharmacology of drugs, because the compounds are classified as inactive (if not surpass the barrier) and active (if it exceeds the barrier), i.e., inactive compounds avoid the side effects [23] . In Table 4 are shown the penetration values of blood-brain barrier, and classified according to Ma et al. [24] , compounds which exhibit higher values of 1 (CBrain/CBlood > 1) are considered
![]()
Table 3. Absorption properties of the modified compounds.
[a]percentage of human intestinal absorption; [b]cell permeability (Caco-2 in nm/s); [c]cell permeability Maden Darby Canine Kidney in nm/s; [d]skin permeability.
![]()
Table 4. Distribution properties in percentages of PPB and penetration of the blood brain barrier for 15 compounds selected from the BindingDB.
active in the CNS may cause collateral effects, and compounds that have values below 1 (CBrain/CBlood < 1) are classified as inactive in the CNS. Therefore, inhibitors analyzed showed that all the compounds have less penetration values than 1, and ranged from 0.0411815 to 0.481764, being that compound 1 showed a value of CBrain/CBlood = 0.135467.
In Table 5 are shown the results of distribution properties for the proposed inhibitors, and these modified compounds (A, B and C) were considered poor plasma proteins binding with values of 42.041932%, 72.714481% and 25.975504%, respectively. Compound B showed a higher strength in plasma protein binding relative to compound 1, having a variation between them ±1.489344. However, compound B presented higher half-life than that compound 1, according to Godin (1995), because the connected portion acts as a reservoir where the drug is released slowly so that it remains in equilibrium with unbound fraction that is being metabolized and excreted [19] [20] . The proposed inhibitors, Table 5, showed penetration values less than 1, i.e., they are classified as inactive in the CNS by presenting low absorption in comparing its precursor compound (compound 1, Figure 1) that presented an absorption of 0.135467.
The Ames test assesses mutagenicity of the compounds, and this test uses Salmonella typhimurium strains with modifications in genes responsible for the histidine synthesis, because this histidine is important for growth. The test assesses the ability of the mutagenic agent to cause growth inhibition in medium without histidine [25] . Inhibitors (1 - 15) submitted to this test showed positive prediction, i.e., were predicted as a mutagen, and only compound 3 showed a negative prediction, being predicted as non-mutagenic.
The carcinogenicity test has the objective of identifying the tumorigenic potential in animals and risk assessment in humans. Such test requires much study time (>2 years) and the preADMET online server predicts the results from data NTP (National Toxicology Program) and EUA/FDA, because they are data made in vivo in rats and mice for 2 years [26] . Study performed by Vieira et al. (2014), used computational methods to classify the degree of anticancer activity against a cell line of human hepatocellular carcinoma (HepG2) of artemisinin derivatives, based on their molecular structure it is concluded that such derivatives have anticancer effects, showing satisfactory results in predicting of pharmacokinetic and toxicological properties using preADMET server [27] . When analyzing carcinogenicity in mice the compounds 1 - 12, 14 and 15 were predicted as negative, i.e., have carcinogenic evidence and compound 13 has positive prediction, i.e., show non-carcinogenic evidence for mice.
By analyzing rat carcinogenicity the compounds 1 - 6 and 9 - 15 were predicted positive showing non-carci- nogenic evidence, and compounds 7 and 8, presented negative predictions showing strong carcinogenic evidence, according Table 6. To the proposed compounds A, B and C were predicted as mutagenic and presented negative predictions, i.e., carcinogenic both to rats and mice, as shown in Table 7.
![]()
Table 5. Distribution properties of the modified compounds.
![]()
Table 6. Toxicological properties of mutagenicity (Ames test) and carcinogenicity (mouse and rat) of selected compounds from the BindingDB.
![]()
Table 7. Toxicological properties of mutagenicity (Ames test) and carcinogenicity (mouse and rat) of the modified compounds.
4. Conclusions
Pharmacokinetic and toxicological properties (ADME/Tox) of the 15 most active inhibitors of deoxyhypusine synthase target selected database BindingDB showed satisfactory results for oral administration, because it have been adequate to the Lipinski parameters. However, the modifications performed from the most active compound, compound 1, not obtained results that enabled administration by the same via, because HIA these decreased having less values than 31.686200%. Being that human intestinal absorption is the sum of absorption and bioavailability, assessed from the proportion of excretion or cumulative excretion in urine, bile and feces.
In relation to the penetration of the blood-brain barrier (CBrain/CBlood) inhibitors exhibited penetration values less than 1, being classified as inactive in the CNS. The modified compounds have decreased penetration CBrain/ CBlood, and showed lower values when compared to compound 1 which had value of CBrain/CBlood = 0.135467, thus being able to have less side effects. However, the same drug did not present an absorption level equal to or higher when compared to the precursor compound, modifications showed positive and satisfactory results and depending on the administered did it will produce the same effects with degree of adverse effects on the CNS.
Acknowledgements
We gratefully acknowledge the support provided by the Brazilian Agency National Council of Scientific and Technological Development (CNPq-Brazil). The authors would like to thank the Scientific Initiation Program (IC/CNPq/UNIFAP), and the Laboratory of Modeling and Computational Chemistry of Federal University of Amapá for computational support.
NOTES
*Corresponding author.