1. Introduction
Cyclodextrins (CDs) [1] [2] are a family of cyclic oligomers, composed of 1-4 linked β-D-glucose units, which includes three industrially produced native CDs: α-CD (six glucose units), β-CD (seven units), and γ-CD (eight units) as well as several other oligosaccharides having more glucose units. They are crystalline, homogeneous, non-hygroscopic and hollow truncated cone-shaped molecules with all the secondary hydroxyl groups [O(2)-H and O(3)-H] crowning the wider rim, while all the primary hydroxyl groups [O(6)-H] located on the narrower rim. CD exterior, containing numerous hydroxyl groups, is fairly hydrophilic, while its cavity, which contains two rings of methine protons, is relatively hydrophobic.
Enormous interest in CDs is due to their unique structural topography which enables them to form inclusion complexes with a wide variety of hydrophobic guest molecules [3] . The host-guest complexation exerts a profound effect on the physicochemical properties of the guest such as solubility, stability, and taste modification by masking off unpleasant flavors and odors etc. Hence, CDs are an important attribute in industrial applications such as pharmaceutical, food, cosmetics, packing and textile industries [4] .
CDs are considered as good excipients for drug formulation and since complexation of drug with CD results in enhanced stability, water solubility, and modified pharmacokinetics of the drugs, with subsequent reduction in adverse effects, these complexes are of particular interest in the area of pharmaceutical sciences [5] -[7] . Moreover, CD complexes are considered good models to understand molecular and chiral recognition mechanism which is the basic concept to understand biological assemblies and their functions which attracts researchers to their structural studies [8] .
Several techniques are employed to characterize inclusion complexes in solution. For instance, the interaction between drugs and cyclodextrins in solution has been demonstrated by phase solubility curves [9] , UV-visible and fluorescence spectroscopy [10] , induced circular dichroism and microcalorimetry [11] , among others [12] . However, these methods provide only indirect information about the inclusion phenomenon and little or no structural information about the complex. NMR spectroscopy not only provides direct evidence to the formation of inclusion complex, but also provides information useful for detailed structure elucidation of the complex [13] .
We have studied complexation of benzhexol with β-CD in aqueous medium by NMR spectroscopy and computer simulation method. Benzhexol, an anti-Parkinsonian agent of the anti-muscarinic class, is white crystalline powder, slightly soluble in water [14] . It was selected because phenyl ring is considered to best fit for β-CD cavity. The aim of the study was to see whether careful interpretation of a poorly resolved ROESY (Rotating-frame Overhauser Effect SpectroscopY) spectrum, which can be generally used only to confirm inclusion of the guest into cavity, can provide more structural details in combination with simulation studies [15] [16] .

Benzhexol
2. Experimental Section
All NMR spectra were recorded at room temperature on a JEOL 500 MHz instrument in D2O. No external reference was used and HDO signal at 4.800 ppm was used as internal reference. 1H NMR spectra were recorded for pure β-CD, pure benzhexol and their six mixtures having molar ratios [H/G] from 0.25 to 2.00. Samples were prepared by dissolving 9 mg of β-CD and calculated amounts of benzhexol in 0.5 ml of D2O.
COrrelation SpectroscopY (COSY) spectrum for 1:1 benzhexol-β-CD mixture was acquired using gradient- selected COSY, utilizing pulsed field gradients in 1:1 ratio with a relaxation delay of 1.5 s. Acquisition parameters consisted of 1280 data points in F2 over a spectral width of 9000 Hz with an acquisition time of 0.109 s and 256 data points in F1 domain covering a spectral width of 7500 Hz taking acquisition time of 0.034 s. ROESY spectrum for 1:1 benzhexol-β-CD mixture was recorded under spin lock condition by acquisition of 1024 (F2) × 55 (F1) complex data points, sweeping over same spectral width in both dimensions as described in COSY. ROESY data was acquired in phase sensitive mode with a mixing time of 0.5 s, spin lock power of 21 dB, and a relaxation delay of 1.5 s.
Molecular mechanics studies were performed in vacuum at 298 K using Allinger’s MM2 force field as included in CS Chem3D Pro (Cambridge Soft Corp). Previously published neutron diffraction coordinates of hydrated β-CD were used, after removal of water coordinates, as starting coordinates [17] . Structure of benzhexol was drawn using CS Chem3D Pro and its geometry was then minimized to a root mean square (RMS) value 0.1 kcalmol−1A−1. MM2 minimization studies were carried out by keeping the optimized benzhexol molecule into the β-CD cavity followed by geometry optimization at the corresponding level of calculation.
3. Results and Discussion
3.1. 1H NMR Spectroscopic Studies
Direct evidence for the formation of inclusion complexes is obtained by simply comparing the 1H NMR data of pure host and guest with that of a mixture of host and guest [18] . There are several reports on the NMR studies of CDs inclusion complexes and it is considered best technique for such studies in solution [19] [20] . Chemical shift changes (Δδ) for the protons of both host as well as guest are studied. When an aromatic guest is accommodated within the CD cavity, there is generally an upfield shift in CD cavity proton resonances while guest protons exhibit concomitant downfield shift changes. These shift changes are attributed to the ring current effect of the aromatic guest in the cavity.
An unambiguous 1H NMR assignment of CD as well as guest proton resonances, especially aromatic proton of the guest, in pure form as well as in the mixture is the foremost requirement for such studies. 1H NMR spectrum of pure benzhexol exhibited phenyl proton signals as two multiplets centered at 7.38 integrating for two protons (H-2,6) and at 7.46 integrating for three protons (H-3,4,5). The aliphatic proton resonances were observed as several signals in the region 0.90 - 3.45 which totally integrated for 25 protons which were well accounted. 1H NMR assignment of β-CD is known but was confirmed with the help of 2D COSY spectral data, just to avoid any ambiguity, and was found in good agreement to the reported. In the 1H NMR spectra of mixtures of benzhexol and β-CD, the signals for the two components did not interfere with each other and were easily identified by COSY data. Phenyl protons of benzhexol were observed as three signals in the presence of β-CD: a doublet at 7.41 (H-2,6), a triplet at 7.46 (H-4), and a doublet of triplet at 7.55 (H-3,5). All the β-CD proton resonances in the presence of benzhexol were identified by COSY data.
A cursory examination of spectra of pure benzhexol, pure β-CD and a mixture of these two showed that signals for β-CD protons situated in the cavity (H-3’ and H-5’) shifted highfield in the presence of benzhexol compared to pure β-CD. There was a slight highfield shift in the H-6’ signal while H-2’ moved slightly lowfield but signal for H-4’ was not at all affected. Similarly, proton resonances of phenyl ring exhibited significant changes in the presence of β-CD. There were downfield shifts in the H-2,6 and H-3,5 protons resonances while H-4 proton signal did not show any significant shift (Figure 1). Chemical shift changes were also observed in few aliphatic protons signals. These observations clearly indicated the formation of benzhexol-β-CD complex in solution by the penetration of phenyl ring of benzhexol into the β-CD cavity.
![]()
![]()
Figure 1. Part of spectra of (a) pure β-CD and BEN:β-CD mixture and (b) pure BEN and BEN:β-CD mixture showing shift changes.
3.2. Stoichiometry and Binding Constant of Complex
Stoichiometry and binding constant of the complex were determined by NMR titration method. NMR spectra of six mixtures of benzhexol and β-CD having molar ratios were recorded. Samples were prepared by dissolving constant amount of β-CD and calculated amounts of benzhexol in D2O. Titration yielded shift change data (Δδ)
for both the cavity protons which was directly related to the concentration of benzhexol. The data was used to determine stoichiometry and binding constant of the complex using Scott’s equation [21] which for a 1:1 benzhexol-β-CD complex can be written as:


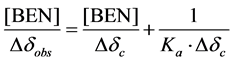

where [BEN] is the molar concentration of benzhexol, Δδobs is the chemical shift difference observed for β-CD proton resonances for a given benzhexol concentration, Δδc is the difference in chemical shift between the complexed and the free β-CD at saturation and Ka is binding constant. The ΔδH-3’ and ΔδH-5’ data was plotted against concentration of benzhexol (Table 1) in the form of [BEN]/Δδobs versus [BEN] which gave excellent linear fits confirming 1:1 stoichiometry of the complex (Figure 2). The binding constant was calculated to be 59.3 M−1.
![]()
Figure 2. Scott’s plot for 1:1 BEN-β-CD complex.
3.3. ROESY Spectral Studies
ROESY provides information about through space correlations [22] . Mixing time is an important parameter to obtain high resolution ROESY spectra. More ROEs are observed for longer mixing time and large molecules but, using longer mixing time produces spin diffusion and both COSY and TOCSY artifacts are observed due to coherence transfer between scalar-coupled spins [23] . A mixing time of 0.2 - 0.25 s is suitable for obtaining high resolution spectra (no spin diffusion) of CD complexes but several hours are required for acquisition. On the other hand, a spectrum can be recorded in few minutes with longer mixing time, e.g. 0.5 s, but will be poorly resolved.
ROESY spectrum of mixture of β-CD and benzhexol was recorded with a mixing time of 0.5 s. An intramolecular peak of benzhexol between aromatic signal at 7.41 and aliphatic signal at 2.50 confirmed our assignment
to peak at 7.41 to H-2,6 protons. Prominent intermolecular cross peaks were observed between cavity protons and phenyl protons which confirmed the position of phenyl ring into β-CD cavity. No crosspeak between para proton and cavity protons was observed (Figure 3). Generally, no more information about the structure of complex is inferred from such a ROESY spectrum.
ROESY peak intensities depends mainly on interproton distances but are also affected by several other factors. It has been shown, that all these factors being equal for two pairs of spins in a given experiment, ratio of peak intensities is equal to the ratio of inverse sixth power of the distances of interacting spins [24] . To see whether any information about the exact orientation of phenyl ring in the cavity can be obtained, relative ROESY peak intensities were estimated for various complexes. Two modes of entry from both cavity openings at various depths were analyzed for relative peak intensities (Figure 4).
All possible complexes by mode I was ruled out because they must show crosspeak between para-phenyl and cavity protons. The expected relative peak intensities for various complexes by mode II are shown in Table 2. As can be seen, WS II and NB II complexes can only explain observed relative intensities in the spectrum which suggest that phenyl ring must be positioned close to wider rim. Since NB II complex is not feasible due to steric hindrance, phenyl ring must enter from wider side.
![]()
Figure 3. Parts of ROESY spectrum showing intermolecular crosspeaks.
![]()
Figure 4. Two modes of entry from both cavity openings at various depths.
3.4. Molecular Mechanics (MM) Studies
Energy minimization studies were performed for all the possible complexes using Allinger’s force field [25] . Complexes were obtained by manually placing the phenyl ring in desired positions into β-CD cavity followed by energy minimization. The steric energies for all the studied complexes are given in Table 3.
The results show that complexes obtained by wide side entry were more stable. The most stable complexes
were obtained when the phenyl ring was placed in the center of the cavity from wider. But on minimization, phenyl ring moved slightly outward and acquired a position which was intermediate of the modes I and II (Figure 4). The position of phenyl ring in cavity was identical in both these conformations (Figure 5). Thus it can be assumed, on the basis of their steric energies, that lower energy conformation (97.8436 kcalmol−1) is the most probable structure of benzhexol-β-CD complex.
![]()
Table 2. Observed and expected relative ROESY peak intensities.
![]()
Table 3. Steric energies (kcalmol−1) obtained by MM2 energy minimization studies.
![]()
Figure 5. Side and top views of two lowest energy conformations. Mode I (WC): ESteric = 98.4268 kcalmol−1. Mode II (WC): ESteric = 97.8436 kcalmol−1.
4. Conclusion
The three-dimensional structure of benzhexol-β-CD complex in aqueous medium was established by NMR spectroscopic and computational methods. The formation of 1:1 inclusion complex was confirmed by NMR titration method. ROESY spectrum recorded with a mixing time of 0.5 s only confirmed the entry of phenyl ring into β-CD cavity. Analysis of all the possible conformations for their relative crosspeak intensities clearly indicated entry of phenyl ring from wider side of cavity and its position near wider rim. These conclusions were well supported by molecular mechanics studies. The results show that valuable structural information can be obtained by studying the ROESY peak intensity ratios even from a poorly resolved spectrum which can be recorded in few minutes.
Acknowledgements
Shazia Shamim is thankful to UGC, Government of India, for the financial assistance.
NOTES
*Corresponding author.