A Cell-Penetrating Peptide for Inhibiting MAPKAP Kinase 2-Mediated Inflammatory Cytokine Release Following Glial Cell Activation ()
1. Introduction
During the brain’s initial injury response, the blood-brain barrier is broken, resulting in hypoxia, injury, and eventual death of neuronal and glial cells [1] [2] . Blood-borne macrophages and injured cells initialize a cascade of signals that induce microglial activation at the injury site within 24 hours [3] -[5] . Once activated, microglia releases nitric oxide at an injury site, resulting in the formation of reactive oxygen species, and subsequently neuronal cell death [6] . Activated microglia also releases inflammatory cytokines, such as tumor necrosis factor- α (TNF-α), colony stimulating factors, and members of the interleukin (IL) family, which contribute to the cell proliferation of reactive microglia and activate astrocytes [4] [5] [7] -[9] . This response also creates chronic local inflammation that contributes to the degeneration of neurons [10] -[12] . The ability to control microglial activation is of significant importance to a variety of applications, such as neuroprostheses, whose functional lifespan is limited by glial cell activation [13] . Therefore, strategies must be developed to mitigate glial cell activation and neuroinflammation.
One possible way to reduce glial cell activation and neuroinflammation is through the down-regulation of inflammatory cytokines. While there is evidence that overexpression of cytokines, such as IL-1β, IL-6, and TNF-α, leads to a more intense secondary injury response [14] -[17] , complete depletion of cytokine expression can lead to neuronal apoptosis [18] [19] . Therefore, down-regulating, but not eliminating, expression of selective cytokines is a possible method to mitigate glial cell activation.
One enzyme involved in inflammatory cytokine release is mitogen-activated protein kinase-activated protein kinase 2 (MK2), which plays a direct role in inflammatory cytokine upregulation in the brain [20] . Following brain inflammation, p38 MAP kinase activates MK2, resulting in an increase in cytokine mRNA biosynthesis, stability, and translation [21] -[24] . However, microglia from MK2 deficient mice shows suppressed p38 and heat shock protein 27 (HSP27) phosphorylation following activation [25] . Inflammatory cytokine production in these mice was also reduced to basal levels, but not completely eliminated [25] . MK2 deficient microglia/neuron co-cultures also showed reduced neurotoxicity following microglial activation, resulting in increased neuronal viability [20] [25] [26] . Therefore, MK2 inhibition may be a possible method to reduce inflammatory cytokine expression following brain injury, resulting in reduced glial cell activation and increased neuron viability.
Protein kinase regulation can be accomplished through the use of substrate-based inhibiting peptides [27] . The peptide sequence, KKKALNRQLGVAA, was identified as a substrate-based inhibitor of MK2 in vitro [28] . Cell-penetrating peptides (CPPs) have also been studied as a mechanism for delivering MK2-inhibiting peptide sequences into cells. Lopes et al. (2009) demonstrated that the CPP, WLRRIKAWLRRI conjugated to KALNRQLGVAA, inhibited MK2 activity as determined by reduced HSP27 phosphorylation [29] . This CPP competes with substrates, such as heterogeneous nuclear ribonucleoprotein A0 (hnRNPA0) [30] and tristetraprolin (TTP) [31] , at the MK2 active site, resulting in a reduction in substrate phosphorylation. Ward et al. (2009) found the peptide sequence, YARAAARQARAKALARQLGVAA (MK2i), to be a non-toxic and more specific inhibitor of MK2 [32] [33] . Therefore, it is hypothesized that MK2i will reduce the expression of inflammatory cytokines from activated microglia, resulting in less secondary damage to the cortical environment. This report provides in vitro evidence of MK2i’s ability to lower inflammatory cytokine production, decrease cortical cell death, and reduce glial cell activation following TNF-α and lipopolysaccharide (LPS) treatment within a mixed primary cortical cell culture.
2. Materials and Methods
2.1. Primary Cell Culture
Embryonic rat cortical tissue was obtained from E17 Sprague-Dawley rats immediately following removal of embryos from a pregnant rat’s abdomen or was purchased from BrainBits, LLC (Springfield, IL). This tissue was placed in a 50 mL conical tube containing 5 mL of Solution 1 (NaCl 7.24 g/L; KCl 0.4 g/L; NaH2PO4 0.14 g/L; Glucose 2.61 g/L; Hepes 5.96 g/L; MgSO4 0.295 g/L; Bovine Serum Albumin 3 g/L) or 5 mL of Hibernate E (BrainBits, Springfield, IL). Under sterile conditions, 18 µL of trypsin solution (7.5 mg/mL in 0.9% saline) was added and tissue was passed through a 5 mL pipet several times to disassociate tissue. After the conical tube was placed in a 37˚C water bath for 20 minutes, 100 µL of trypsin inhibitor/DNAse solution (2.5 mg/mL trypsin inhibitor, 400 µg/mL DNase in 0.9% saline) was added to the tube and the tissue was pipetted several times with a 5-mL pipet. Tissue was centrifuged at 1000 rpm for 5 minutes at room temperature and supernatant was poured off. Cells were re-suspended in 16 mL of Hibernate E and 100 µL of trypsin inhibitor/DNAse solution and pipetted up and down several times. Cells were filtered through a cell strainer and centrifuged at 1400 rpm for 5 min at room temperature. Supernatant was poured off and cells were re-suspended in media. Primary cells were then plated in poly-D-lysine-coated 96-well plates at a seeding density of 625,000 cells/cm2 and incubated for 7 - 9 days at 37˚C, with media being changed every other day. After 7 - 9 days, separate groups of cortical cells were treated with 5 or 10 ng/mL TNF-α or 50 ng/mL LPS + cell media solution, respectively, for 4, 8, and 24 hours to activate microglia. Separate groups of cortical cells were also treated with a 0.5, 1, or 3 mM MK2i + 5 or 10 ng/mL TNF-α or 50 ng/mL LPS + cell media solution for 4, 8, and 24 hours. Additionally, a separate group of cortical cells received only cell media to determine basal cytokine levels. For a given time point, all treatments were conducted on cortical cells from the same embryonic dissection. After treatments, media was removed and stored at −80˚C.
2.2. Immunocytochemistry
Cell media was replaced with phosphate-buffered 4% formaldehyde solution (pH 7.4) for 5 minutes, followed by three washes using HEPES-buffered Hanks’ saline containing 10 mg/L sodium azide; pH 7.4 (HBHS). A Triton X-100 (Sigma-Aldrich, St. Louis, MO) solution (0.25% by volume in HBHS) was applied for 10 minutes, followed by three washes in HBHS. Blocking solution, consisting of 10% normal goat serum in HBHS (V/V), was then applied for 30 minutes, followed by three washes with HBHS. Microglia, astrocytes, and neurons were labeled using antibodies against ionized calcium binding adaptor molecule 1 (Iba1) (019-19741, Wako Pure Chemical Industries, Osaka, Japan), glial fibrillary acidic protein (GFAP) (AB5541, Millipore, Temecula, CA), and β-3-tubulin (β-3-tub) (MMS-435P, Covance, Princeton, NJ), respectively. Antibodies were applied for 2 hours, at a dilution of 1:400 in HBHS. After two 10 minute washes and one 30 minute wash, secondary antibodies were applied. Alexa fluor 488 goat anti-mouse, Alexa fluor 555 goat anti-chicken, and Alexa fluor 635 goat anti-rabbit (all from Invitrogen) were applied at 1:400, along with Hoechst 33,342 at 1:10,000 (Invitrogen, Eugene, OR), in HBHS for 2 hours. All immunocytochemistry procedures were performed at room temperature. Cells were stored away from light at 4˚C in HBHS prior to imaging.
2.3. Cell Imaging
Cell imaging was accomplished using an Olympus IX81 microscope equipped with Olympus FV1000 laser confocal system, through Olympus 10×/0.40 air and 40×/0.80 water emersion objectives (Olympus America, Center Valley, PA). Image channels were collected sequentially using 488 nm, 543 nm, and 633 nm laser lines (CVI Melles Griot, Carlsbad, CA), along with a tunable MaiTai laser (Spectra-Physics/Newport, Santa Clara, CA) set to 740 nm. Two-photon excitation of Hoechst 33,342 was driven by this 740 nm excitation, and the emission was collected by an external PMT (R3896, Hamamatsu, Bridgewater, NJ) equipped with a 405/40 nm filter (Chroma, Rockingham, VT). Internal detectors collected all other channels. A wide aperture setting (400 µm) was used to capture representative images from the labeled cultures; laser power and PMT voltage settings were held constant. Related image-channel levels were set equal, allowing visual comparison of fluorescent intensity, using Olympus Fluoview V1.7 software. Microglia cell number quantification was accomplished using the CellCount for the public domain program ImageJ (National Institutes of Health, Bethesda, MD), with two 1272 × 1272 µm images counted per treatment. Fluorescent intensity quantification was performed by measuring mean intensity across 800 × 800 pixel (1272 × 1272 µm) raw images (two per treatment) using the analysis function in ImageJ, then averaging the mean intensities within each treatment. Scale bars were added and figure layouts were designed using Photoshop CS2 (Adobe Systems, San Jose, CA). Statistical analysis was conducted using Statistical Analysis Software (SAS, Cary, NC) version 9.2.
2.4. IL-6 ELISAs
IL-6 ELISAs were conducted to quantify changes in inflammatory cytokine production using the Rat IL-6 ELISA Development Kit (Peprotech Inc., Rocky Hill, NJ). 100 µL of 1 µg/mL antigen-affinity purified goat anti-rat IL-6 was added to ELISA microplate wells (NuncMaxisorp, Rochester, NY) and incubated at room temperature overnight. Wells were washed 4 times with 300 µL wash buffer (0.05% Tween-20 in 1x phosphate buffered saline (PBS)). 300 µL block buffer (1% BSA in 1x PBS) was added to each well and incubated for 1 hour at room temperature. Block buffer was aspirated and wells were washed 4 times with wash buffer. 8 ng/mL recombinant IL-6 was then diluted to zero in diluent (0.05% Tween-20, 0.1% BSA in 1x PBS). 100 µL of diluted recombinant IL-6 solutions were added in triplicate into microplate wells. 100 µL of media from all treatments were also added to microplate wells in sextuplicate. Microplates were then incubated at room temperature for 2 hours, aspirated, and washed 4 times with wash buffer. 100 µL of 0.25 µg/mL biotinylated antigen-affinity purified goat anti-rat IL-6 was added to microplate wells and incubated for 2 hours. Microplates were aspirated and washed 4 times with wash buffer. 6 µL Avidin Peroxidase 1:2000 was diluted in 12 mL diluent. 100 µL of this solution was added to wells and incubated for 30 minutes at room temperature, aspirated, and washed 4 times with wash buffer. 100 µL of 2,2’-Azino-bis-(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) Liquid Substrate Solution (Sigma-Aldrich, St. Louis, MO) was added to each well. Absorbance was then measured using a Spectramax M5 Microplate Reader (Molecular Devices, Sunnyvale, CA). Absorbance values were monitored at 5-minute intervals for 50 minutes.
2.5. IL-1β ELISAs
IL-1β ELISAs were conducted to quantify changes in inflammatory cytokine production using the Rat IL-1β ELISA Development Kit (Peprotech Inc., Rocky Hill, NJ). Methods were identical to those of the IL-6 ELISA assays. Absorbance values were monitored at 5-minute intervals for 25 minutes.
2.6. Additional IL-6 and IL-1β Assays
Cytokine assays were conducted to quantify changes in inflammatory cytokine production using Proinflammatory Panel 1 (rat) Kits (Meso Scale Discovery, Rockville, MD). 150 µL of Blocker H was added into each well and shaken at 700 rpm for 1 hour. The 96-well MSD plate was then washed three times with 150 µL of wash buffer (1x PBS + 0.05% Tween 20) per well and 50 µL of calibrator or sample solution was added into each well of the plate. The plate was then sealed with adhesive tape and shaken at 700 rpm for 2 hours at room temperature. After three washes with wash buffer, the detection antibody was then added (25 µL/well) and the plate was again sealed and shaken at 700 rpm for 2 hours. After three more washes with wash buffer, 150 µL of 2× read buffer was added to each well and the plate was analyzed using Sector Imager 2400A (Meso Scale Discovery, Rockville, MD). Both IL-6 and IL-1β were examined in this study and data were analyzed using MSD Discovery Workbench Software.
2.7. Live-Dead Assay
Live-dead assays were conducted using the Molecular Probe LIVE/DEAD® Viability/Cytotoxicity Kit for Mammalian Cells to quantify cell death in cortical cell cultures following all treatments. A sample size of six was used for each treatment. Prior to experiments, optimal concentration of the live cell dye, Calcein-AM (CA), and the dead cell dye, Ethidium-1 (EthD-1), was determined to be 6 µM. Also, optimal time for dye incubation was 30 minutes.
One-half hour before the 24-hour time point, untreated cultures were killed with a 30-minute 70% ethanol treatment, then washed twice with 250 µL 1x PBS. One group of control dead cells received 100 µL of the 6 µM CA solution to determine the background fluorescence of CA. A second group of control dead cells received 100 µL of the 6 µM EthD-1 solution to determine the maximum fluorescence for EthD-1. Conversely, one group of control live cells received 100 µL of the 6 µM EthD-1 solution to determine the background fluorescence of EthD-1, while a second group of control live cells received 100 µL of the 6 µM CA solution to determine the maximum fluorescence for CA.
At the 24 hour time point, cultures were washed twice with 250 µL 1x PBS. Then, 100 µL of a 6 µM EthD- 1/6 µM CA working solution was added to each treated well. Fluorescence was then measured using a Spectramax M5 Microplate Reader (Molecular Devices). EthD-1 required an excitation wavelength of 530 nm and an emission wavelength of 645 nm. CA required an excitation wavelength of 485 nm and an emission wavelength of 530 nm.
2.8. Ramification Index
The following equation was used to calculate the ramification index (RI) of individual microglia [34] :
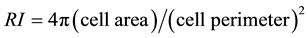 (1)
(1)
Cell images were obtained as previously described and, using Image J, microglia imaged from each treatment well was carefully traced. Subsequently, Image J was used to calculate cell area and cell perimeter for each tracing. These values were then utilized to obtain an average RI for each treatment group using the above equation.
2.9. Western Blot Analysis
Cortical cells were grown for 1 week in 30 mm Petri dishes, followed by 24-hour MK2i treatment. Media was removed and cells were rinsed two times with ice-cold Tris-Buffered Saline (TBS). 100 µL of lysis buffer (0.54 g/mL 9M Urea, 0.04 g/mL CHAPS, 10 µL Sigma Phosphatase Inhibitor Cocktail 1/mL) was added to dishes and cells were scraped to a corner of the dish using a cell scraper. Cells were added to a chilled, 1.5 mL LoBind protein tube (Sigma Aldrich, St. Louis, MO) and frozen at −80˚C. Thawed cells were vortexed using the Disruptor Genie (Scientific Industries #SI D236, Bohemia, NY) for three hours at 4˚C, then centrifuged at 20, 160 Relative Centrifugal Force (RCF) for 20 minutes at 4˚C. Supernatant was collected into a LoBind tube and frozen at −80˚C. The bicinchoninic assay (BCA) Protein Assay Kit (Pierce #PI23225) was used to run a total protein assay on thawed supernatant. Protein standards were made using bovine serum albumin (BSA). 25 µL of each standard and sample were pipetted in triplicate into a 96-well microplate. 200 µL of working reagent was added to each well and mixed thoroughly on a plate shaker for 30 seconds. The microplate was then covered, incubated at 37˚C for 30 minutes, and cooled at room temperature. The absorbance of each well was then measured at 562 nm using a Spectramax M5 Microplate Reader (Molecular Devices, Sunnyvale, CA).
Following the protein assay, 15 µg of total protein was added to aliquot tubes and diluted with 50 µL Beta Mercaptoethanol (Bio-Rad #161-0710, Hercules, CA) and 950 µL Laemmli Sample Buffer. Tubes were heated for 5 minutes at 100˚C, vortexed, and centrifuged. 2 µL of the solution was then loaded into Tris-hydrochloride (Tris-HCl) Bio-Rad gel lanes, and the gel was run. Polyvinylidenefluoride (PVDF) membranes (Bio-Rad #162- 0174) were soaked in methanol for 1 - 2 minutes in preparation for gel transfer. Gels were then transferred to the PVDF membrane using a Trans-Blot SD System (Bio-Rad #1703848) and placed into a blocking buffer solution (Odyssey, #927-40000) in a cold room overnight on a rocking platform (VWR, Rocking Platform Model 200). The membrane was washed twice with TBS, with each wash preceding a 15-minute incubation on the rocking platform.
Primary antibodies for Beta Actin (Abcam-ab8226, Cambridge, England) and MK2 (Abcam-ab32567) were added to a 5 mL solution of TBS, 0.05% Tween-20, and 0.25% BSA. The solution and membrane were placed in a plastic pouch, sealed and incubated on a rocking platform overnight at 4˚C. The membrane was removed and rinsed four times with a 1× TBS +0.05% Tween-20 solution. Secondary antibodies were then added to a 5 mL solution of TBS, 0.05% Tween-20, and 0.25% BSA. The solution and membrane were placed in a plastic pouch, sealed and incubated in the dark on a rocking platform for 1 hour at room temperature. The membrane was removed, rinsed four times with a 1× TBS +0.05% Tween-20 solution, and washed with 1× TBS for 15 minutes on a rocking platform to remove residual Tween-20. The membrane was then incubated in stripping buffer (15 g glycine, 1 g SDS, 10 mL Tween-20) for 10 - 15 minutes, rinsed with 1× TBS +0.05% Tween-20 for 5 minutes, and rinsed with TBS. Protein-antibody complexes were visualized using an Odyssey Imaging System. Complexes were quantified by measuring the intensity of each blot and normalizing MK2 protein expression to the β-actin control.
2.10. Data Analysis
Data analysis for all experiments was conducted with Statistical Analysis Software (SAS, Cary, NC) version 9.2. First, Dunnett’s method for multiple pairwise comparisons was used to determine the effect of TNF-α and LPS treatments on IL-6 concentration (pg/mL) compared to untreated cells (control). Control sample means were compared to sample means from 5 ng/mL and 10 ng/mL TNF-α and 50 ng/mL LPS treatments. No significant difference in IL-6 concentration was shown when comparing untreated controls to 5 or 10 ng/mL TNF-α treatments and therefore, 5 or 10 ng/mL TNF-α treatments were used as two controls for two separate sets of experiments. However, a significant difference in IL-6 concentration was shown when comparing untreated controls to 50 ng/mL LPS treatments and therefore, untreated cells were used as a control in those experiments. Dunnett’s method for multiple pairwise comparisons was then used to compare the effect of TNF-α + MK2i treatment on IL-6 concentration to the TNF-α controls. Sample means from 5 ng/mL TNF-α treatment were compared to sample means following 5 ng/mL + 0.5 mM MK2i, 5 ng/mL + 1 mM MK2i, and 5 ng/mL + 3 mM MK2i treatment. Similarly, sample means from 10 ng/mL TNF-α treatment were compared to sample means following 10 ng/mL + 0.5 mM MK2i, 10 ng/mL + 1 mM MK2i, and 10 ng/mL + 3 mM MK2i treatment. Dunnett’s method for multiple pairwise comparisons was also used to compare the effect of LPS + MK2i treatment on IL-6 concentration to the untreated controls. Sample means from untreated controls and 50 ng/mL LPS treat- ment were compared to sample means following 50 ng/mL + 0.5 mM MK2i and 50 ng/mL + 1 mM MK2i treatment. IL-1β ELISA analysis was identical to IL-6 ELISA analysis.
Next, Dunnett’s method for multiple pairwise comparisons was used to determine the effect of TNF-α treatment on the percentage of dead cortical cells (%Dead) compared to untreated cells (control). Control sample means were compared to sample means from 5 ng/mL and 10 ng/mL TNF-α treatments. No significant difference in %Dead was shown when comparing untreated controls to 5 or 10 ng/mL TNF-α treatment. Therefore, 5 or 10 ng/mL TNF-α treatments were used as two controls for two separate sets of experiments. Dunnett’s method for multiple pairwise comparisons was then used to compare the effect of TNF-α + MK2i treatment on %Dead to the TNF-α controls. Sample means from 5 ng/mL TNF-α treatment were compared with sample means following 5 ng/mL + 0.5 mM MK2i, 5 ng/mL + 1 mM MK2i, and 5 ng/mL + 3 mM MK2i treatment. Similarly, sample means from 10 ng/mL TNF-α treatment were compared to sample means following 10 ng/mL + 0.5 mM MK2i, 10 ng/mL + 1 mM MK2i, and 10 ng/mL + 3 mM MK2i treatment.
Next, Dunnett’s method for multiple pairwise comparisons was used to determine the effect of TNF-α treatment on microglia cell number compared to untreated cells (control). Control sample means were compared to sample means from 5 ng/mL and 10 ng/mL TNF-α treatments. No significant difference in microglia cell number was shown following treatment with 5 or 10 ng/mL TNF-α. Therefore, 5 or 10 ng/mL TNF-α treatments were used as two controls for two separate sets of experiments. Dunnett’s method for multiple pairwise comparisons was then used to compare the effect of TNF-α + MK2i treatment on microglia cell number to the TNF-α controls. Sample cell number means from 5 ng/mL TNF-α treatment were compared to sample means following 5 ng/mL + 0.5 mM MK2i, 5 ng/mL + 1 mM MK2i, and 5 ng/mL + 3 mM MK2i treatment. Similarly, sample means from 10 ng/mL TNF-α treatment were compared to sample means following 10 ng/mL + 0.5 mM MK2i, 10 ng/mL + 1 mM MK2i, and 10 ng/mL + 3 mM MK2i treatment. Β3-tubulin, GFAP, and Iba1 fluorescence analysis was identical to Iba1 cell number analysis.
3. Results
3.1. Confirmation of Triple (Mixed) Cell Cultures
Immunocytochemical images confirm the presence of microglia (Iba1), neurons (β-3-tub), and astrocytes (GFAP) in 1-week old mixed cortical cultures (Figure 1). While other cortical cells may exist in these cultures, we clearly identify the existence of the cells necessary for this study.
3.2. MK2i Significantly Reduces Mean IL-6 and IL-1β Concentration Following Pro-Inflammatory Stimulation
Dunnett’s method was used to compare the effect of TNF-α + MK2i treatment on IL-6 concentration to TNF-α treatmentalone. For the 5 and 10 ng/mL TNF-α controls, analysis showed that mean IL-6 concentrations were significantly lowered when TNF-α treatment was combined with 0.5, 1, or 3 mM MK2i for 4, 8, or 24 hours (Figures 2(a)-(c)).
Dunnett’s method was also used to compare the effect of TNF-α + MK2i treatment on IL-1β concentration to
![]() (a)
(a)![]() (b)
(b)
Figure 1. 1 week mixed cortical culture image under (a) 10× and (b) 40× objectives; immunostained for neurons (β-3-tub), astrocytes (GFAP), microglia (Iba1), and cell nuclei (Hoechst 33,342) within the same culture (overlay). Brightfield transmission of light (Trans) is also shown. Scale bar is 100 µm in (a) and 50 µm in (b).
TNF-α treatmentalone (Figures 2(d)-(f)). For the 5 ng/mL TNF-α controls, mean IL-1β concentrations were significantly lowered when TNF-α treatment was combined with 0.5, 1, or 3 mM MK2i for 4, 8, and 24 hours. For the 10 ng/mL TNF-α controls, mean IL-1β concentrations were significantly lowered when TNF-α treatment was combined with 0.5, 1, and 3 mM MK2i for 4 and 8 hours. After 24 hours, mean IL-1β concentrations were also lowered when TNF-α treatment was combined with 0.5, 1, and 3 mM MK2i; however, only 3 mM MK2i produced a statistically significant decrease. It should be noted that the concentrations of TNF-α used in these experiments were chosen for their ability to upregulate inflammatory cytokine expression as lower concentrations of TNF-α showed no significant effects (data not shown).
Dunnett’s method was used to compare the effect of LPS + MK2i treatment on IL-6 and IL-1β concentrations to untreated controls and LPS treatment. Results were similar to TNF-α stimulated cultures as LPS combined with 0.5 and 1 mM MK2i lowered IL-6 and IL-1β concentrations compared to LPS treatment alone; however, only a select number of specific time points and concentrations showed a statistically significant decrease (data not shown). The ability of MK2i to downregulate cytokine concentration following LPS stimulation is consistent with our previously published work [35] .
3.3. MK2i Significantly Increases Cortical Cell Viability
Dunnett’s method was used to compare the effect of TNF-α + MK2i treatment on the percentage of dead cortical
![]()
Figure 2. Mean IL-6 and IL-1β concentrations (in pg/mL) following 4, 8, and 24 hour treatments with TNF-α or TNF-α + MK2i. IL-6 (a)-(c): For the 5 and 10 ng/mL TNF-α controls, mean IL-6 concentrations were significantly lowered when combined with 0.5, 1, or 3 mM MK2i treatment for 4, 8, or 24 hours; IL-1β (d-f): For the 5 ng/mL TNF-α controls, mean IL-1β concentrations were significantly lowered when TNF-α treatment was combined with 0.5, 1, or 3 mM MK2i for 4, 8, and 24 hours. For the 10 ng/mL TNF-α controls, mean IL-1β concentrations were significantly lowered when TNF-α treatment was combined with 0.5, 1 or 3 mM MK2i treatment for 4 and 8 hours. After 24 hours, mean IL-1β concentrations were also lowered when combined with 0.5, 1, or 3 mM MK2i treatment; however, only 3 mM MK2i produced a statistically significant decrease, which may be due to the toxicity of this high concentration. *: p < 0.05; **: p < 0.005; ***: p < 0.001; ****: p < 0.0001.
cells to TNF-α treatment. In Figure 3, %Dead values for TNF-α + MK2i treatments were compared to TNF-α only treatments. After 24 hours, 5 ng/mL TNF-α + MK2i treatment significantly decreased %Dead cells. 10 ng/ mL TNF-α + MK2i treatment also decreased %Dead values; however, this decrease was not statistically significant.
3.4. Assessment of MK2i-Induced Changes in Microglia and Neurons
Untreated microglia displayed an amoeboid or round morphology with high Iba1 intensity in the center of the
![]()
Figure 3. %Dead indicates the percentage of dead cells per well and illustrates differences in cell viability when cells are treated with TNF-α + MK2i vs. TNF-α treatment alone (24 hours). 5 ng/mL TNF-α + MK2i treatment results in a significantly lower percentage of dead cortical cells than 5 ng/mL TNF-α treatment alone. 10 ng/mL TNF-α + MK2i treatment also results in a lower percentage of dead cortical cells than 10 ng/mL TNF-α treatment alone; however, this difference was not statistically significant. *: p < 0.05; **: p < 0.005; ***: p < 0.001.
cell (Figure 4(a)). Treatments of 5 and 10 ng/mL TNF-α resulted in spreading of microglial processes (Figure 4(b)), a sign of embryonic microglial activation [36] . MK2i treatment alone caused microglia to swell and spread Iba1 intensity throughout the cell while maintaining its amoeboid morphology (Figure 4(c)). Treatments of TNF-α + 0.5 or 1 mM MK2i suppressed microglial spreading caused by TNF-α treatments, and maintained cell swelling seen in MK2i only treatments (Figure 4(d)).
To further quantify these microglia observations, the Ramification Index (RI) was calculated and used to compare TNF-α + MK2i treatments to untreated controls and TNF-α only treatments. 5 and 10 ng/mL TNF-α treatments significantly lowered RI compared to untreated controls, indicating a more branched appearance (Figure 5). And while not statistically significant, we noted that microglia treated with TNF-α + MK2i had a higher RI, indicating a more amoeboid or round morphology, when compared to cultures treated with TNF-α alone (Figure 5).
Treatments of 5 and 10 ng/mL TNF-α resulted in increased neuronal cell death while treatments of TNF-α + 0.5 or 1 mM MK2i suppressed the neuronal cell death (Figure 6 and Figure 7). However, treatment of TNF-α + 3 mM MK2i resulted in increased cell death as well as various areas of intense β3-tubulin (Figure 6 and Figure 7), suggesting a possible toxic response to high levels of MK2i.
Analysis of image data showed no significant differences in Iba1, β3-tubulin, or GFAP fluorescence intensity following 24-hour TNF-α + MK2i treatments (data not shown). There was also no significant change in microglia cell number following 24-hour TNF-α + MK2i treatments (data not shown). These results confirm a similar concentration of cell-type specific proteins within each culture following various treatments.
3.5. MK2i Does Not Directly Inhibit MK2 Expression
Western blot analysis determined the effect of the MK2i peptide on MK2 protein expression. Comparison of control blots to treatment blots showed no significant reduction in MK2 expression following 24 hour 0.5 mM MK2i treatment (Figure 8). These results support MK2i’s proposed mechanism of action that MK2i interferes with substrate phosphorylation downstream to MK2, but does not reduce MK2 expression itself.
4. Discussion
MK2i is a novel type of cell-penetrating peptide (CPP). CPPs can be utilized to deliver bioactive molecules into cells. Frankel and Pabo’s discovery of the TAT HIV protein’s ability to be rapidly taken up by cells launched a
![]()
Figure 4. Typical microglia morphology displayed in our treated and untreated culture wells is shown here with antibody labeled Iba1. Tracings of representative microglia are shown on right. (a) Prior to treatment, microglia typically displayed an amoeboid or rounded morphology with high Iba1 intensity in the center of the cell. (b) TNF-α treatment resulted in spreading of microglial processes, a sign of embryonic microglial motility and activation. (c) MK2i treatment caused microglia to swell, and spread Iba1 intensity throughout the cell while maintaining its amoeboid morphology. (d) TNF-α + MK2i treatment suppressed the microglial spreading seen in TNF-α only treatments, but swelling caused by MK2i only treatments remained. Scale bar is 25 µm.
![]()
Figure 5. Mean ramification index (RI) for microglia following 24 hour treatments with TNF-α or TNF-α + MK2i. 5 ng/mL TNF-α (#) and 10 ng/mL TNF-α (*) treatments had a significantly lower RI when compared to untreated controls. TNF-α + MK2i treatments had a higher RI than TNF-α treatments alone, but these results were not statistically significant.#, *: p < 0.05.
great interest into the therapeutic use of TAT to deliver compounds across the cell membrane [37] . Through crosslinking to the compound to be delivered, TAT has the ability to deliver molecules into cells in vitro [38] . With MK2i, the CPP domain, YARAAARQARA, is used to deliver the MAPKAP kinase 2 (MK2)-inhibiting peptide, KALARQLGVAA, into cells in vitro. It has been shown that certain CPPs, if not designed properly, can alone inhibit kinases and that modifying the sequence of a kinase-inhibiting peptide can alter the kinase specificity [33] . Therefore, we were not able to create a scrambled peptide sequence that was inactive. Using the novel MK2i peptide, we were able to reduce MK2-mediated inflammatory cytokine production, microglial activation, and cortical cell toxicity following TNF-α and LPS treatments. The presented results demonstrate that treatment concentrations of 0.5 and 1 mM MK2i are effective at down-regulating, but not eliminating, inflame-
![]()
Figure 6. TNF-α treatments of 5 and 10 ng/mL resulted in morphological changes in neurons resembling neuronal cell damage, which was suppressed with 0.5 and 1 mM MK2i. However, this suppression was not maintained at 3 mM MK2i treatment, which also resulted in cell damage. Scale bar is 100 µm, all cells labeled with β3-tubulin.
![]()
Figure 7. Neurons following 24-hour treatments. 5 and 10 ng/mL TNF-α treatments resulted in increased neuronal cell death and was shown to be toxic to dendrites (arrowheads). This neuronal cell death caused by TNF-α was suppressed by 0.5 and 1 mM MK2i treatments. However, 3 mM MK2i treatment seems to have an adverse effect on the neurons and may indicate a potentially toxic dose (arrows). Scale bar is 50 µm.
![]()
Figure 8. Western blot analysis confirms that MK2 activity is maintained following 24-hour MK2i treatment (Molecular Weights: β-action = 42 kDa; MK2 = 46 kDa).
matory cytokine production without causing neuronal cell damage after microglial activation in vitro. The images of microglia, probed with Iba-1, and the calculated ramification index show that MK2i suppresses microglial spreading induced by treatment with TNF-a. This is consistent with the understanding that non-activated embryonic microglia are naturally amoeboid in shape and branch out when they mature or become activated [36] . Additionally, IL-1β has been shown to be a marker of microglia activation [39] and therefore, its down regulation further supports the claim that MK2i suppresses microglial activation. The overall increase in cortical cell viability (Figure 3) coupled with the images of the neural population (Figure 6) suggest that there is a neuronal protective effect to treating with MK2i. MK2i treatment creates a more viable cellular environment, potentially through avoiding TNF-a-induced neuroinflammation [40] -[42] . While 3 mM MK2i treatment also lowered inflammatory cytokine production, it produced significant neuronal cell damage, thus demonstrating an upper effective concentration limit. Additionally, we showed that MK2i did not directly inhibit MK2 expression (Figure 8) as expected from its proposed mechanism of action. However, we were unable to demonstrate a decrease in downstream substrate phosphorylation because of a lack of specificity of the rat antibodies, but Brugnano et al. (2011) showed that MK2i downregulated HSP27 phosphorylation in human THP-1 monocytes [35] . These results show the promise of MK2i as a therapeutic to inhibit glial activation after CNS trauma, such as that which results upon implantation of neuroprosthetic devices. In order to utilize the MK2i peptide to inhibit the reactive tissue response to intracortical microelectrodes, it may be necessary to coat the peptide onto microelectrodes prior to implantation. Silicon based microelectrodes coated with thin-film, silica sol-gel coatings have been shown to maintain their electrical properties after chronic implantation [43] . Therefore, thin-film, silica sol-gel coatings are a possible mechanism for MK2i treatment at implantation site.
While the use of MK2i focuses on inhibiting inflammatory cytokine release from microglia, there are other mechanisms that may play a role in the neuronal cell death and glial cell activation following a brain injury. The release of the interferon-γ cytokine from injured neurons introduces β-Amyloid, which induces microglia nitric oxide production. This cascade of reactions inhibits respiratory enzymes, oxidizes the SH group of proteins, and enhances DNA injury, resulting in neuronal cell death [44] . Injured neurons are also unable to upregulate cytosolic or mitochondrial superoxide dismutase enzymes, which are important for neuronal resistance to oxidative stress [45] .
Activated microglia also expresses a variety of chemokines that play a role in migration and communication [46] . The activated microglia has chemokine receptors that contribute to microglial proliferation, migration, and cytoskeletal reformation [42] . The receptors also allow activated microglia to communicate with injured neurons or dying cells through chemoattractants, such as chemoattractant protein-1 [4] and fractalkine [47] [48] .
Microglia and astrocytes also have Toll-like receptors (TLRs) that recognize host molecules from damaged tissues [42] [49] . TLR ligation of heat-shock proteins [50] [51] , heparin sulfate [52] , necrotic cell molecules [53] [54] , and bacterial cells [55] -[57] result in the upregulation of inflammatory target genes that encode chemokines, cytokines, and enzymes [42] [49] [58] [59] . The ligation of bacterial cells is important, due to the high prevalence of implantable device-associated bacterial infections, which remains a major concern to healthcare facilities worldwide [60] . Therefore, it is possible that bacteria may be introduced into the brain during microelectrode implantation, playing a contributing role in glial cell activation through TLRs. While MK2i will help in suppressing glial activation, it may need to be coupled with additional therapeutics to mitigate other injury responses and promote the healthiest healing environment.
5. Conclusion
Through in vitro studies, we demonstrate the ability of a novel cell-penetrating peptide (MK2i) to down-regulate, but not completely eliminate MK2-mediated cytokine expression following glial cell activationin mixed cortical cell cultures. MK2i was also shown to increase cell viability, reduce microglial activation and elicit a possible neural protective effect. Given the critical role of cytokine production in chronic neuroinflammation and glial scar formation, these results demonstrate the potential of MK2i as a therapeutic to reduce glial activation following CNS trauma, such as that which occurs upon implantation of neuroprosthetic devices.
Acknowledgements
The authors would like to thank the Purdue Research Foundation, the Indiana Spinal Cord and Brain Injury Research Board (grant #00014975), and the National Science Foundation (NSF CCLI-0728668) for their financial support, as well as the Weldon School of Biomedical Engineering and the Purdue Graduate School.
NOTES
*J. R. Wodicka and N. I. Onunkwo contributed equally to this work.
#Corresponding author.