1. Introduction
AD (Alzheimer’s disease) is a wide spread brain disease among older people. The disease is characterized by the formation of Amyloid β-peptides (Aβ) in the form of oligomers and fibrillar amyloid deposition resulted after proteolytic cleavage of the amyloid precursor protein (APP) by β- and g-secretases leading to the actual causes of the disease [1] -[4] .
Several studies have suggested that the accumulation of pathogenic amyloid-β (Aβ) assemblies in the brain can result in the dismantling of synapses, neuronal circuits and networks, which are associated with memory loss and cognitive decline [5] - [7] . In addition it has been reported that an imbalance between the formation rate and the clearance rate of Aβ deposits in the brain increases the progression of AD [8] .
Despite the general interest concerning oligomers toxicity compared to fibrils, there is a real need to understand the structure of these insoluble forms of Aβ. Some studies have shown that fibrillar Aβ was organized in a parallel β-sheet conformation while oligomeric Aβ were attributed to an antiparallel β-sheet structure [9] .
Identification of structural changes in the oligomerization process and explanations of the thermodynamics associated with the conformational changes in the assembly of Aβ will facilitate identification of therapeutic agents. Furthermore, the linkage between oligomerization and cellular dysfunction or toxicity is yet not understood.
Recent reports suggested that neurodegenerative disorders, such as Alzheimer’s and Parkinson’s diseases, might be accelerated by anesthesia, proposing that inhaled anesthetics promote protein oligomerization through occupancy of intermolecular cavities, and, in the case of amyloid-β, that these cavities are most prevalent in the diffusible oligomeric precursor of fibrils, hereafter termed intermediate oligomers [10] - [12] .
This study was undertaken to understand the oligomerization process in terms of its causes, structural characteristic, and to suggest mechanisms that may be used by the protein to prevent aggregation. Fourier transform infrared (FT-IR) and atomic force microscopy (AFM) were used to investigate the changes in the secondary structure of beta Amyloid (1-40) due to its interaction with an anesthetic propofol on one side and L-arginine on the other side.
2. Materials and Methods
Aβ (1-40) in powder form, propofol in solution form (5.390 M) and L-arginine in powder form were all purchased from Sigma Aldrich chemical company and used without further purification.
2.1. Preparation of Stock Solutions
An amount of 0.5 mg of Aβ (1-40) was dissolved in 250 μL of triple distilled water to give a concentration of 0.462 mM as a stock solution. A 1.338 mg of L-arginine was dissolved in 2 ml of PBS (PH 7.4) to prepare 3.84 mM L-arginine stock solution. Then different concentrations of L-arginine (3.84, 2.88, 1.92, 1.44, 0.96, 0.72, and 0.48 mM) were prepared. Propofol solutions with concentrations of (3.82, 2.88, 1.92, 1.44, 0.96, and 0.72 mM) were prepared using phosphate buffer saline (PBS) and the original propofol stock solution (5.390 M).
The final solutions of Aβ-propofol were prepared by mixing equal volumes of Aβ and propofol stock solutions. Aβ concentration in all samples was 0.231 mM, and the propofol concentrations in the final protein-drug solution were (1.92, 1.44, 0.96, 0.72, 0.48, 0.36, and.0.24 mM). For the final Aβ-L-arginine solution, equal volumes of Aβ and L-arginine stock solutions were mixed to give L-arginine concentrations of (1.92, 1.44, 0.96, 0.72, 0.48, 0.36, and 0.24 mM).
The solutions were incubated for one hour at 20˚C before spectroscopic measurements were taken.
2.2. UV-Absorption Spectra
The absorption spectra were obtained by using a NanoDrop ND-100 spectrophotometer. The spectra were recorded for 1 - 2 μL liquid samples of free Aβ and for its complexes with propofol solutions with the concentrations of (0.36 mM, 0.48 mM, 0.72 mM, 0.96 mM, 1.44 mM, and 1.92 mM). The same measurements were taken for Aβ-L-arginine complexes with similar concentrations. Repeated measurements were done for all the samples and no significant differences were observed.
2.3. Fluorescence Spectroscopy
The fluorescence measurements were performed by a NanoDrop® ND-3300 Fluorospectrometer at 25˚C. The excitation source comes from one of three solid-state light emitting diodes (LED’s). The excitation source options include: UV LED with maximum excitation 365 nm, Blue LED with excitation 470 nm, and white LED from 500 - 650 nm excitation. A 2048-element CCD array detector covering the range of 400 - 750 nm is connected by an optical fiber to the optical measurement surface. The excitation is done on the wavelength of 350 nm and the maximum emission wavelength is at 431 nm.
2.4. FTIR Spectroscopic Measurements
The FT-IR measurements were obtained on a Bruker IFS 66/S spectrophotometer equipped with a liquid nitrogen-cooled MCT detector and a KBr beam splitter. The spectrometer was continuously purged with dry air during the measurements. Samples were prepared after two hours of incubation for the Aβ-propofol and Aβ- L-arginine solutions at room temperature, four drops of each sample were placed on a certain area on a silicon window plate and left to dry at room temperature. The hydrated films on one side of a silicon window plate of the samples containing different concentrations of propofol or L-arginine with the same protein content.
The absorption spectra were obtained in the wave number range of 400 - 4000 cm−1. A spectrum was taken as an average of 60 scans to increase the signal to noise ratio, and the spectral resolution was at 4 cm−1. The aperture used in this study was 8 mm, which was found that to gives the best signal to noise ratio. Baseline correction, normalization and peak areas calculations were performed for all the spectra by OPUS software. The peak positions were determined using the second derivative by OPUS software.
The infrared spectra of Aβ-propofol complex were obtained in the featured region of 1800 - 1000 cm−1. The FT-IR spectrum of free Aβ was acquired by subtracting the absorption of the buffer solution from the spectrum of the protein solution. For the net interaction effect, the difference spectra {(protein solution + propofol solution) ? (protein solution)} were generated using the featureless region of the protein solution 1800 - 2200 cm−1 as an internal standard [13] . The accuracy of this subtraction method is tested using several control samples with the same protein or drug concentrations, which resulted into a flat base line formation. The spectral differences were used to investigate the nature of the Aβ-ligand interaction.
For the temperature dependence studies, ligand-protein complexes on silicon windows were placed into an infrared cell window. The temperature in the cell was controlled by an external water path which was increased gradually from 20˚C to 80˚C at 3˚C per 10 minute scan rate.
2.5. AFM Measurements
The prepared stock solutions of Aβ-propofol complex were incubated for 2 hrs at room temperature. A 10 µl of the solution was placed on certain area of a freshly cleaved mica substrate, and left to dry at room temperature. Free Aβ, Aβ-propofol and Aβ-L-arginine solutions were characterized by AFM equipped with 4 × 4 µm2 piezoelectric scanner. Analyses were carried out using tapping mode at room temperature using AFM cantilevers with a spring constant 2 N/m (Micro cantilever, OLYMPUS). All scanned images were analyzed using WSxM 5.0 Develop 3.2 software.
3. Results and Discussion
3.1. UV-Absorption Spectroscopy
The Aβ-propofol and Aβ L-arginine binding constants were determined using UV absorption spectroscopy as reported for several ligand-protein complexes [14] - [16] . The absorption spectra for different concentrations of propofol and L-arginine in Aβ are shown in Figure 1. The excitation has been done at 210 nm, while the absorption is recorded at 280 nm. The UV absorbance intensity of Aβ has increased with increasing propofol concentration, while it has decreased with increasing L-arginine concentration.
The absorption data was treated using linear reciprocal plots based on the following equation.
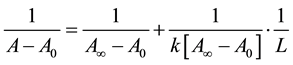 (1)
(1)
where A0 corresponds to the initial absorption of protein at 280 nm in the absence of ligands, A∞ is the final absorption of the protein ligand complex, and A is the recorded absorption at different propofol concentrations (L). The double reciprocal plot of 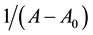 vs.
vs. 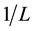 is linear (Figure 2) and the binding constant (K) for Aβ-
is linear (Figure 2) and the binding constant (K) for Aβ-
![]() (A)
(A)![]() (B)
(B)
Figure 1. UV absorbance spectra of Aβ-propofol (A) and of Aβ-arginine (B) at the following concentrations for both cases (a: 0.0 mM, b: 0.36 mM, c: 0.48 mM, d: 0.72 mM, e: 0.96 mM, f: 1.44 mM, and g: 1.92 mM).
propofol is calculated to be 2.13 × 102 M−1, and for Aβ-L-arginine complexes is calculated to be 0.35 × 102 M−1. The binding constant for Aβ-L-arginine is lower than that of Aβ-propofol. Drugs with low binding constants are known to be more effective in their interactions with protein [17] .
The UV absorbance intensity of Aβ has increased with increasing propofol concentration, while it has decreased with increasing L-arginine concentration. The increase of band intensity due to the increase of propofol concentration can be related to an increase in the number of absorption sites as a result of structural changes of the Aβ-propofol complex. The reason for the decrease of intensity in Aβ-L-arginine can be attributed to the weak stability due to the presence of mainly hydrogen bonding between protein donor atoms and the ligands polar groups or an indirect ligand-protein interaction through water molecules [18] .
3.2. Fluorescence Spectroscopy
The intrinsic fluorescence In Aβ (1-40) is mainly due to tyrosine residue. This is because no tryptophan is found in the structure of Aβ (1-40) and because phenylalanine has very low quantum yield [19] [20] . The fluorescence spectra of Aβ free and at various concentrations of propofol (0.24, 0.36, 0.48, 0.96, 1.44, and 1.92 mM) are shown in (Figure 3(A)). The emission fluorescence spectra of Aβ with various L-arginine concentrations (0.24, 0.36, 0.48, 0.96, 1.44, and 1.92 mM) are shown in (Figure 3(B)). The excitation has been performed at 350 nm while the emission occurred at 431 nm.
The fluorescence intensity of Aβ decreases regularly with the increase of ligand’s concentration. Other factors can affect fluorescence intensity of a compound such as molecular rearrangements, exited state reactions, energy transfer, ground state complex formation, and collisional quenching [21] - [23] . The Dynamic quenching process can be described by the Stern-Volmer equation [24] - [26]
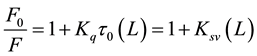 (2)
(2)
where F and F0 are the fluorescence intensity with and without quencher, Kq is the quenching rate constant of the biomolecule, Ksv is the Stern-Volmer quenching constant, t0 is the average lifetime of the biomolecule without quencher, and (L) is the concentration of propofol or L-arginine. As can be seen from Figure 4, the Stern- Volmer plots are linear and the slopes are equal to the Stern-Volmer quenching constants for Aβ-propofol and Aβ-L-arginine complexes with these values: 1.21 × 103 L∙mol−1 and 1.49 × 102 L∙mol−1 respectively. Fluores-
![]()
![]() (A) (B)
(A) (B)
Figure 3. The fluorescence emission spectra of Aβ with various concentrations of propofol in (A) and with various concentrations of L-arginine in (B), the concentration are: (a = 0.0, b = 0.24, c = 0.36, d = 0.48, e = 0.96, f = 1.44, and g = 1.92 mM).
![]()
![]() (A) (B)
(A) (B)
Figure 4. The plot of ![]() vs L for Aβ-propofol in (A) and for Aβ-L-arginine in (B).
vs L for Aβ-propofol in (A) and for Aβ-L-arginine in (B).
cence quenching can be induced by different mechanisms, which are usually classified into dynamic and static quenching. Dynamic quenching arises from collisional encounters between the fluorophore and quencher, and static quenching results from the formation of a ground state complex between the fluorophore and the quencher [27] .
The quenching rate constants for Aβ-propofol and Aβ-arginine were then calculated and found to be 1.1 × 1012 L∙Mol−1∙s−1 and 1.35 × 1012 L∙Mol−1∙s−1 respectively. The obtained values of the quenching rate constants for propofol and L-arginine are larger than the maximum dynamic quenching constants for various quenchers with biopolymers (2 × 1010 L∙Mol−1∙s−1) which confirms that static quenching is dominant in these complexes [28] [29] . When the static quenching equation is used [30] .
![]() (3)
(3)
where K is the binding constant of the ligand with Aβ. The value of K can be determined from the slope and the y-intercept of the straight line in Figure 5. The two binding constants are found to be 2.81 × 102 M−1 for Aβ- propofol and 0.37 × 102 M−1 for Aβ-L-arginine which agrees well with the values obtained earlier by UV spectroscopy and supports the effective role of static quenching.
The highly effective quenching rate constants in these cases have led to a lower value of binding constants between the ligands and Aβ due to an effective hydrogen bonding between the ligands and Aβ.
Fluorescence quenching is attributed to changes in the environment of the protein fluorophores caused by the presence of the ligand [31] and is likely to be associated with amyloid fibrils [32] .
3.3. FTIR Spectroscopy
The FTIR absorption spectra in the amide I and amide II regions are taken for different concentrations of Aβ- propofol and Aβ-L-arginine samples. The analysis of the secondary structure of Aβ in the amide I which occurs between 1600 cm−1 and 1700 cm−1 yields several bands. The peaks of these bands correspond to the C=O stretching vibrations of the amide group, coupled to the C-N stretching and C-C-N deformation mode [28] . The peaks in the amide II (1600 - 1480) cm−1 region are due to the coupling of the N-H in-plane bending and C-N stretching modes [33] .
The difference spectra of [(Aβ + propofol or L-arginine) − Aβ] were obtained to show the intensity variation, and the results are shown in (Figure 6 and Figure 7). For the [(Aβ + propofol) − Aβ] case, Figure 6 shows a strong positive feature at 1630 cm−1 in the amide I region in addition to another weak positive feature at 1670 cm−1. For amide II region there is a strong positive feature at 1550 cm−1. These features were obtained at propofol concentrations of (0.48, 0.72, and 1.44 mM).
For [(Aβ + L-arginine) − Aβ] interaction, Figure 7 shows the difference spectra for Aβ with different L-argi- nine concentrations. In the amide I region there is a strong negative feature at 1629 cm−1, and another weak negative feature at 1668 cm−1, while in amide II region there is a strong negative feature at 1538 cm−1 and another weak negative feature at 1493 cm−1.
It is clearly shown in Figure 6 that the strong positive features in amide I and amide II regions become stronger as the propofol concentration increased. On the other hand the strong negative features in amide I and amide II regions become stronger with increasing L-arginine concentration.
The observed positive features are attributed to the increase in the intensity of the amide I bands at 1630, and 1670 cm−1, and amide II band at 1550 cm−1 as a result of propofol interaction with Aβ. This increase in the intensity is due to the increase in the number of β-sheet bonds [26] . The negative featured noticed in the difference spectra of [(Aβ-L-arginine) − Aβ] are due to the decrease in the intensity with increasing L-arginine concentration. This decrease in the intensity is due to the decrease in the number of β-sheet bonds.
![]()
![]() (A) (B)
(A) (B)
Figure 5. The plot of ![]() vs 1/L for Aβ-propofol is shown in (A) and for Aβ-L-arginine is shown in (B).
vs 1/L for Aβ-propofol is shown in (A) and for Aβ-L-arginine is shown in (B).
![]()
![]() (A) (B)
(A) (B)
Figure 6. (A) FTIR spectra for Aβ free and Aβ-propofol complex (1.44 mM), and (B) the difference spectra of Aβ free and Aβ-propofol at different concentrations.
![]()
Figure 7. (A) FTIR spectra for Aβ free and Aβ-L-arginine complex 0.72 mM, and (B) the difference spectra of Aβ free and Aβ-L-arginine at different concentrations.
The component bands of the amide I and amide II regions were determined using Fourier self-deconvolution (FSD) and second derivative resolution with curve fitting procedures. This procedure has provided a basis for the quantitative estimation of protein secondary structure [27] [30] [34] and [35] .
Based on several previous proteins’ studies [9] [19] , the amide I bands are assigned as follows: (1600 - 1612 cm−1) to anti-parallel β-sheet, (1612 - 1630 cm−1) to parallel β-sheet, (1630 - 1650 cm−1) to random coil, (1650 - 1670 cm−1) to α-helix, (1670 - 1685 cm−1) to β-turns, and (1685 - 1700 cm−1) to anti-parallel β-sheet. Similarly for the amide II region, the absorption spectra is composed of six bands assigned in the following order: (1480 - 1500 cm−1) to parallel β-sheet, (1500 - 1520 cm−1) to random coil, (1520 - 1538 cm−1) to anti parallel β-sheet, (1538 - 1560 cm−1) to α-helix, (1560 - 1580 cm−1) to β-turns, and (1580 - 1600 cm−1) to anti-parallel β-sheet.
In this work a quantitative analysis of the protein secondary structure for Aβ free, Aβ-propofol and Aβ-L-ar- ginine complexes in dehydrated films are determined from the shape of amide I and amide II bands. Figure 8 shows the spectra and its second derivative for the amide I and the amide II regions. The curve-fitted graphs for the amide I and amide II regions are shown in Figure 9 for Aβ-propofol complex and in Figure 10 for Aβ-L-
![]()
Figure 8. (A) the second derivative spectra of Aβ free, and the spectra of both Aβ-propofol (B) and Aβ-L-arginine (C) at the following concentrations (a = 0.0 mM, b = 0.48 mM, c = 0.72 mM, d = 1.44 mM, and e = 1.92 mM).
arginine complex. The curve fitted graphs show the secondary structure determinations of the Aβ free and Aβ- propofol and Aβ-L-arginine complexes.
The exact frequency of the vibrations depend on the nature of the hydrogen bonding involving the amide group, and by the particular secondary structure adopted by the protein.
Table 1 listed the peak positions of the separate components of the amide I and amide II regions using both second derivative and FSD procedures. The quantitative estimation of protein secondary structure is based on the assumption that the total absorption due to the carbonyl (C=O) vibration can be represented by the sum of the individual absorptions due to the vibrations of the separate elements of the band [27] .
The percentages of each secondary structure of the Aβ-propofol and Aβ-L-arginine complexes were calculated from the integrated areas of the component bands in amide I and amide II respectively. The relative intensity of each secondary structure component of Aβ before and after the interaction with propofol and L-arginine at different concentrations is shown in Table 2 and Table 3.
The changes in band intensities as a result of changing propofol and L-arginine concentrations while keeping Aβ concentration constant in the amide I and amide II regions are shown in Figure 11. These changes show a decrease in the parallel β-sheet intensity percentage accompanied with an increase in anti-parallel β-sheet structures. This behavior is believed to be caused by the misfolded protein in the presence of propofol as a result of H-bonding formation between propofol and Aβ [34] . The parallel β-sheet has a weak structural arrangement, because the geometry of the individual amino acid molecules forces the hydrogen bonds to occur at angles. Introducing non-planarity in the inter-strand hydrogen bonding make them longer and thus weaker. Contrarily, in the anti-parallel arrangement the hydrogen bonds are aligned directly opposite to each other allowing the inter-
![]()
Table 1. Bands assignment in the absorbance spectra of Aβ with different propofol and L-Arginine concentrations for Amide I - II regions.
![]()
![]()
Table 2. Secondary structure determination for Amide I and Amide II regions in Aβ and its propofol complexes.
![]()
Table 3. Secondary structure determination for Amide I and Amide II regions in Aβ and its L-arginine complexes.
strand hydrogen bonding between carbonyl group and the hydrogen atoms of the amines groups to be planar making a planar inter-strand hydrogen bonding and resulting in a shorter (stronger) and more stable bonds [36] - [38] .
The observed increase in the intensity of anti-parallel β-sheet band as a result of Aβ interaction with propofol is an indication of oligomers or fibrils formation [19] [39] . These changes in intensities can also be noted directly from the fitted curves in (Figure 9). Previous studies using circular dichroism (CD), FTIR spectroscopy, and X-ray diffraction studies confirmed that amyloid fibrils have a β-pleated sheet structure [40] .
The analysis of the absorption spectra of Aβ-L-arginine complexes in amide I and amide II, both parallel and anti-parallel β-sheet intensities shows a gradual decrease with increasing L-arginine concentration as shown in (Figure 11(C), and Figure 11(D)). This decrease is an indication of L-arginine effect in decreasing oligomerization by converting the insoluble (β-sheet rich) fibrils or oligomers into more soluble entities.
3.4. Atomic Force Microscopy (AFM)
AFM technique has been used as an additional method to investigate the effect of propofol and L-arginine on Aβ by taking images for Aβ-free solution, Aβ-propofol complex, and Aβ-arginine complex as shown in (Figures 12(A)-(C) respectively). The heights of the clusters in each of these complexes by scanning the surface of the
samples of Aβ-free, Aβ-propofol, and Aβ-L-arginine are shown respectively to the right of each image.
It is clear from AFM images that Aβ specimens have clustered in a certain area as a result of its interaction with propofol, while in the case of Aβ-L-arginine interaction showed little or no change in cluster formation. From the graph shown in (Figure 12(A)) the height of the clusters in Aβ-free sample was found to be between (30 - 35 nm), while for Aβ-propofol sample it has been found to be (250 - 300 nm) as shown in (Figure 12(B)) and for Aβ-L-arginine sample, the height has been found to be (25 - 50 nm) as shown in (Figure 12(C)).
These results clearly indicate the effect of propofol in increasing the sizes of the oligomers which are shown as spherical shaped objects or clusters. This confirms the results already found by FTIR spectroscopy that propofol enhances oligomers formation.
A little change in the heights of Aβ complex structures are noticed upon interaction with L-arginine as shown in (Figure 12(A), and Figure 12(C)) meaning, L-arginine interacts with Aβ in a way different from that of propofol. These results support the results already found by FTIR spectroscopy.
4. Concluding Remarks
The formation of oligomers in the precursor protein induces structural changes leading to a disturbance in the primary function of the protein. Although the molecular detail underlying formation and growth of amyloid oligomers has eluded structural approaches at this time, exact causes are unrevealed. One possible scenario is to assume that information and images are saved in memory at a particular structural state associated with a certain frequency. When a certain electrical signal is provided by neurons an electromagnetic field is produced in the protein which can be reproduced at any later time if the same electrical signal is provided. It seems that propofol interacts with Aβ in the brain to produce oligomers which behave similar to polarized dielectric material that is sandwiched between different beta-sheets or within the folded β-sheet to create a capacitor-like structure. Inserting this material between different beta-sheets or within the folded β-sheet will increase the internal energy of the folded arrangement leading to a more stable complex. Anesthetic molecules are small, hydrophobic molecules that bind principally in internal protein cavities. The interactions between anesthetic molecules and the cavities within the protein restore Van der Waals contacts and thus provide additional free energy for oligomerization [41] . Similarly the interactions between Aβ and propofol within the β-sheet of the protein can lead to more rigid stable oligomers formations. The resulting solid structure can hinder the reproduction of the original frequencies related to the saved information in the memory of the brain, and prevent electrical and chemical signals’ transmittance which is vital for carrying information among different regions of the brain. Consequently, retrieving related existing images and information becomes very challenging and demanding process. In addition, processing new signals and information may slow down.
The direct cause leading to synaptic and neuronal loss in the brain is not completely clear yet; however, some studies suggest that nerve damage might result from the conversion of nontoxic soluble monomers to non-so- luble toxic oligomers and protofibrils [42] .
Evidence are provided within this manuscript for a novel biophysical interaction leading to structural changes in Aβ-propofol interaction. These changes are evident by the observed variations in the intensities of the absorption spectra of the β-sheet bands in the amide I and amide II regions. Recent NMR investigations of Aβ-propofol interaction reaffirm that smaller molecular sized anesthetics do play a leading role in Aβ oligomerization [43] , and a previous study of pentobarbital-transthyretin interactions have yielded similar results and showed protein oligomerization [28] .
Many studies have proven that amyloid oligomers or fibrils which are found in the brains of patients with Alzheimer disease are rich in β-sheet especially the more stable anti-parallel β-sheet [44] - [46] . Since increasing propofol concentration was shown to increase the intensity of the absorption band of anti-parallel β-sheet, one can deduce that propofol increases the rate of oligomerization or fibrils formation. On the other hand arginine shows an opposite effect as increasing its concentration in Aβ-L-arginine complex causes a decrease in the intensity of anti-parallel β-sheet. It can be concluded that L-arginine not only does not induce oligomerization, but it also may decrease the rates of oligomerization.
In addition, the AFM images did not show any changes with regard to cluster formation for L-arginine interaction with Aβ, while a remarkable increase in heights of the clusters is observed for propofol interaction with Aβ.
While the mechanism of L-arginine’s interaction with Aβ is not yet well understood, it seems to be related to its binding to aromatic groups in partially unfolded states of proteins acting as an effective inhibitory agent for a wide variety of amyloid forming proteins. The strong binding with Aβ peptide monomers results in stable complexes and prevents the formation of soluble oligomers produced by hydrophobic interaction.
Acknowledgements
This work is supported by the German Research Foundation DFG grant No. DR228/24-2.
NOTES
*Corresponding author.