1. Introduction
The step-growth polymerization reaction of bis-trimethylsilyloxyor bis-trimethylsilylthio-phenylenes 1 with activated aryl fluorides 2, the so-called “silyl method”, has found wide application in the synthesis of polyphenylene ether (resp. sulfide) ketones and sulfones (see Scheme 1) [1-13]. It involves nucleophilic aromatic substitution reactions on activated aryl halides e.g. 4,4’-difluorobenzophenone or 4,4’-difluorodiphenyl sulfone 2. The reaction in solution, e.g. in N-methylpyrrolidin-2- one (NMP) as solvent, is usually carried out in the temperature range of 130˚C - 200˚C [1-9]. Simple heating of a bisphenol bis-trimethylsilyl ether with an activated aryl halide does not result in polycondensation reactions [6]. The step-growth polymerization is usually initiated by adding either equimolar amounts of potassium carbonate -typically 1 - 1.5 eq., or catalytic amounts of cesium fluoride—in the range of 0.2 - 3 mol% [1-14,15]. Potassium fluoride was found to be much less active, yielding products of lower inherent viscosity [1-7].
In the context of the synthesis of poly(ether ketone)s, poly(ether sulfone)s and poly(sulfide sulfone)s through step-growth polymerization of activated aryl fluorides with free phenols and thiophenols respectively, it has been reported, that activated aryl ether resp. thioether linkages can be cleaved by fluoride or by water assisted by fluoride [7,12,14-15]. Water might for example be brought into the reaction mixture when the strongly hygroscopic cesium fluoride is used.
A polycondensation reaction applying the “silyl method” is usually formulated as shown in Scheme

Scheme 1. “Silyl-method”-type polycondensation.
1. When performing the reaction in the presence of equivalent amounts of potassium carbonate, equivalent amounts of hexamethyl disiloxane and carbon dioxide as well as two equivalents of potassium fluoride are formed together with the polymer 3 (Scheme 1, method a). In contrast, in the reaction with cesium fluoride, fluorotrim-ethylsilane is formed together with the polymer 3 and regeneration of cesium fluoride from the nucleophilic aromatic substitution step generating a diaryl ether linkage, thus making a small amount of initial CsF sufficient to promote the step-growth polymerization/polyconden-sation (Scheme 1, method b).
2. Results and Discussion
We report here, that the addition of a small amount of the non-toxic and much less hygroscopic cesium carbonate instead of cesium fluoride is sufficient to initiate the polymerization reaction, resulting in the desired stepgrowth polymerization products. The use of small amounts of cesium carbonate instead of the fluoride or equivalent amounts of potassium carbonate is based on the following considerations. In the initial reaction step, cesium carbonate reacts with monomers 1 and 2, resulting in the formation of a small amount of a coupling species 4 together with small amounts of hexamethyldisiloxane, carbon dioxide and cesium fluoride (Scheme 1). Once a small amount of cesium fluoride is formed in situ, it may then act as a catalyst for the main reaction generating a polymer 3 from the starting materials 1 and 2, just like as if a small amount of cesium fluoride would have been added from the start (Scheme 1). The advantages of using cesium carbonate are: less expensive, non-toxic and less hygroscopic than cesium fluoride; the latter being so hygroscopic, that it soon liquefies on the spatula by absorbing moisture from the air during transfer to the reaction vessel.
For the step-growth polymerizations we used N-methylpyrrolidin-2-one as solvent, a temperuture range of 150˚C - 180˚C and added 3 mol% of cesium carbonate as catalyst, in order to assure reaction conditions that allow equilibration reactions and thermodynamic control [7]. Tests with 2-phenylhydroquinone-bis-trimethylsilyl ether 5 and 4,4’-difluorobenzophenone 6 as reactants proved to be successful, and yielded the poly(ether ether ketone) 7 as a polymer with pendant phenyl rings (Scheme 2). The polymer was obtained in 83% yield, inherent viscosity hinh(DMAc, 25˚C, c = 2g×L–1): 0.98 dL×g–1(Table 1). The 1H nmr spectrum of polymer 7 is shown in Figure 1; the signals for proton H-h indicate a 1:1:1:1-mixture of triades resulting from the non-selective orientation of the unsymmetrical monomer 5 in the coupling step. Upon heating the reactants 5 and 6 in the presence of one equivalent of anhydrous potassium carbonate instead, a polymer 7 was obtained in 81% yield with hinh(DMAc, 25˚C, c = 2g×L–1) = 0.48 dL×g–1. A polymer of the same inherent viscosity is obtained, if 1.5 equivalents of potassium carbonate are used. When 1.5 equiv. of cesium carbonate were used instead of potassium carbonate, the data of the resulting polymer 7 were as follows: yield 83%, hinh(DMAc, 25˚C, c = 2g×L–1) = 0.60 dL×g–1 (Table 1). According to spectroscopic data, the polymers are of the same overall structure and are obtained as triadic mixture, no matter whether catalytic or equivalent amounts of cesium carbonate, equivalent amounts of potassium carbonate or catalytic amounts of cesium fluoride were used [10,11,13]. Yields and inherent viscosities are comparable to those reported for structurally related polyetherketones and -sulfones that were prepared via the“silyl-method” using cesium fluoride as catalyst [10,11, 13]. Yields and inherent viscosities are also comparable to polymers that were prepared from free bisphenols and activated aryl fluorides in the presence of either two equivalents of cesium fluoride [16] or one equivalent of sodium carbonate together with catalytic amounts of potassium fluoride [14].
Catalytic amounts of cesium carbonate proved to be sufficient as well for the step-growth polymerization of a bis-trimethylsilyl thioether. Reaction of 4,4’-bis-trimethylsilylthio-diphenyl sulfide 8 with 4,4’-difluordiphenyl sulfone 9 under the same conditions gave the poly (phenylene sulfide sulfone) 10 in good yield (Scheme 3 and Table 1). The 1H nmr spectrum of polymer 10 is shown in Figure 2.

Scheme 2. Cesium carbonate promoted polycondensation to form a poly(ether ether ketone).

Figure 1. 1H nmr spectrum (CDCl3) of Polymer 7, 1:1:1:1- mixture of triades.

Table 1. Yields and viscosities of polymers 7 and 10.
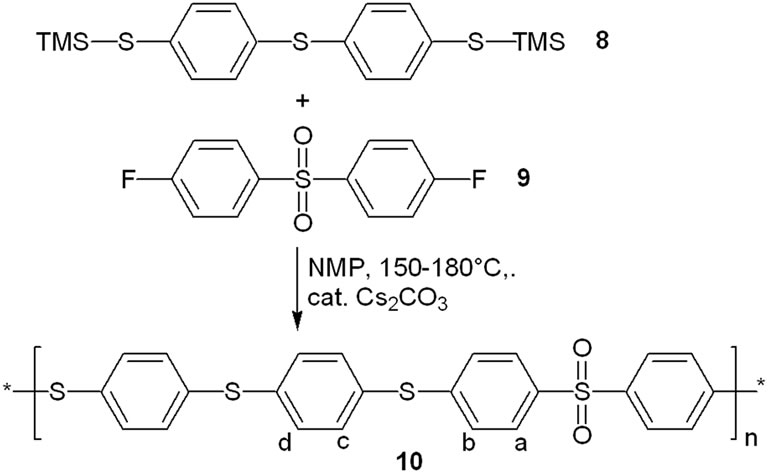
Scheme 3. Cesium carbonate promoted polycondensation to form a poly(sulfide sulfone).

Figure 2. 1H nmr spectrum (CDCl3 + CF3COOD 4:1) of Polymer 10.
Yield and inherent viscosity of the poly(sulfide sulfone) 10 obtained with catalytic amounts of cesium carbonate are well comparable to the product that has been obtained from 4,4’-thio-bis-benzenethiol, 4,4’-difluorodiphenylsulfone and 4 equivalents of cesium fluoride [17]. Yield and inherent viscosities of the polymers prepared here are also well comparable to polyethersulfones as they have been reported very recently [15]. Cesium carbonate has been used as reagent in equimolar amounts alone or 0.5 - 1 equivalents in combination with additional reagents in diaryl ether synthesis and polycondensation reactions [18-25].
3. Conclusion
In conclusion we have found, that catalytic amounts of cesium carbonate are sufficient to initiate the step-growth polymerization of bisphenol bis-trimethylsilyl ethers or bis-thiophenol-bis-TMS-thioethers with activated aryl difluorides under reaction conditions that allow equilibration and thermodynamic control [7]. The addition of equimolar amounts of cesium carbonate is not necessary. The non-toxic, less expensive and less hygroscopic cesium carbonate can thus be used instead of the toxic, expensive and very hygroscopic cesium fluoride.
3.1. Experimental
Materials: 4,4’-Difluorbenzophenone (Lancaster) and 4,4’ -Difluordiphenylsulfone (Sigma-Aldrich) were used as received. 2,5-Bis-trimethylsilyloxybiphenyl, (2-Phenylhydroquinone-bis-TMS-ether) 5, was prepared as reported elsewhere [10,11]. 4,4’-Thio-bisbenzenethiol-bisTMS-thioether (4,4’-Bis-trimethylsilylthio-diphenylsulfide) 8 (mp. 110˚C) was prepared from 4,4’-thio-bis-benzenethiol (Sigma-Aldrich) by a silylation procedure analogous to that described for phenols [10,11] using hexamethyl disilazane as silylating agent. N-Methylpyrrolidin-2-one (abbreviated NMP) (Merck), was dried by distilling from calcium hydride. Cesium carbonate and cesium fluoride (both Sigma-Aldrich) and potassium carbonate (Fluka) were each dried in a vacuum oven prior to use.
Characterization: Solution viscosities were measured in N,N-Dimethylacetamide (DMAc) or N-Methylpyrrolidin-2-one (NMP) as solvent with c = 2 g/l, using an automated Ubbelohde viscosimeter thermostated at 25˚C. Inherent viscosities were calculated from Equation (1) where c is the polymer concentration in g/dL and t and t0 are the running time of the polymer solution and the pure solvent, respectively.
 (1)
(1)
nmr spectra were recorded on a Bruker DRX 500 spectrometer operating at 500.13 MHz for 1H,. 5mm o.d. sample tubes were used. Chloroform-d1 or CDCl3/trifluoroacetic acid (TFA-d) mixtures served as solvent and internal chemical shift reference (CHCl3: 1H 7.26 ppm). Molecular weights were obtained from GPC measurements on a Knauer GPC equipped with Zorbax PSM Trimodal S columns and a RI detector. A mixture of DMAc with 2 vol% water and 3 g/L LiCl was used as eluent. Poly(4-vinyl pyridine) samples served as standards for molecular weight calibration.
3.2. Typical Procedures
Poly(oxy-1,4(2-phenyl)phenyleneoxy-1,4-phenylenecarbonyl-1,4-phenylene) 7 A 50 ml round-bottomed flask is charged with 2.2038 g (10.1 mmoles) of 4,4’-difluorbenzophenone 6, 3.3057 g (10 mmoles) of 2,5-bis-trimethylsilyloxybiphenyl 5 and 16 mL of N-methylpyrrolidin-2-one. The flask is equipped with a reflux condenser, and the reaction mixture is stirred under argon atmosphere at room temperature. After the reactants are completely dissolved, 0.1 g (0.3 mmoles, 3 mol%) of cesium carbonate are added, and the mixture is heated to 150˚C using an oil bath. After stirring the reaction mixture at 150˚C for 20 hours, the oil bath temperature is increased to 180˚C for 2 hours. After cooling to 50˚C the polymer is precipitated by adding the viscous reaction mixture slowly to vigorously stirred ethanol. The polymer is purified by stirring with water followed by ethanol. After drying in a vacuum oven, the yield is 83%. hinh(DMAc, 25˚C): 0.98 dL×g–1. Mw(gpc, DMAc) = 158000 g/mol. 1H NMR (CDCl3): d(ppm) = 7.83, 7.78, 7.72, 7,67 (4d, 4H, J = 7Hz, H-h, 1:1:1:1- mixture of triades), 7.47 (d, 2H, J = 6Hz, H-c), 7.32 (tr, 2H, J = 6Hz, H-b), 7.27 (d, 1H, J = 6Hz, H-f), 7.23 (d, 1H, J = 2Hz, H-d), 7.15 (dtr, 1H, 6Hz, 1.8Hz, H-a), 7.10 (m, 3H, H-e + 2H-g), 6.93, 6.91 (2d pseudo-tr, 2H, J = 7Hz, H-g, 2 isomers of the 1:1:1:1 triadic mixture).
Poly(thio-1,4-phenylenethio-1,4-phenylene-1,4-phenylene-sulfonyl-1,4-phenylene) 10 A 25 ml round-bottomed flask is charged with 3.9477 g (10 mmoles) of bis-TMS-thioether 8, 2.5680 g (10.1 mmoles) of 4,4’-difluordiphenyl sulfone 9 and 18 mL of N-methylpyrrolidin-2-one. The flask is equipped with a reflux condenser, and the mixture is heated under argon atmosphere in an oil bath to 70˚C while being stirred. After the solids have completely dissolved, 0.1 g (0.3 mmoles, 3 mol%) of cesium carbonate is added and the temperature of the oil bath is increased to 150˚C. After stirring the reaction mixture at this temperature for 20 hours, the oil bath temperature is raised to 180˚C and the mixture stirred for an additional 3 hours. After cooling to 50˚C, the polymer is precipitated by adding the viscous reaction mixture slowly to vigorously stirred ethanol. The polymer is purified by stirring with water followed by ethanol. After drying in a vacuum oven, the yield is 82%. hinh(NMP, 25˚C, c = 2g ×L–1): 0.62 dL×g–1. No appropriate solvent found for gpc measurements. 1H NMR (CDCl3 + CF3COOD 4:1): d(ppm) = 7.76 (d, 4H, J = 7 Hz, H-a), 7.47 (d, 4H, J = 7 Hz, H-c), 7.42 (d, 4H, J = 7 Hz, H-d), 7.25 (d, 4H, J = 7 Hz, H-b).
4. Acknowledgements
We thank Dr. H. Komber (IPF) for recording and interpreting the nmr spectra, and Dr. W. Butwilowski (IPF) for determining viscosities. Financial support by the BMBF (Germany)—project 03SF0323A—is gratefully acknowledged.